
Abstract
Transition from compensated to decompensated left ventricular hypertrophy (LVH) is accompanied by functional and structural changes. Here, the aim was to evaluate dystrophin expression in murine models and human subjects with LVH by transverse aortic constriction (TAC) and aortic stenosis (AS), respectively. We determined whether doxycycline (Doxy) prevented dystrophin expression and myocardial stiffness in mice. Additionally, ventricular function recovery was evaluated in patients 1 year after surgery. Mice were subjected to TAC and monitored for 3 weeks. A second group received Doxy treatment after TAC. Patients with AS were stratified by normal left ventricular end-diastolic wall stress (LVEDWS) and high LVEDWS, and groups were compared. In mice, LVH decreased inotropism and increased myocardial stiffness associated with a dystrophin breakdown and a decreased mitochondrial O2 uptake (MitoMVO2). These alterations were attenuated by Doxy. Patients with high LVEDWS showed similar results to those observed in mice. A correlation between dystrophin and myocardial stiffness was observed in both mice and humans. Systolic function at 1 year post-surgery was only recovered in the normal-LVEDWS group. In summary, mice and humans present diastolic dysfunction associated with dystrophin degradation. The recovery of ventricular function was observed only in patients with normal LVEDWS and without dystrophin degradation. In mice, Doxy improved MitoMVO2. Based on our results it is concluded that the LVH with high LVEDWS is associated to a degradation of dystrophin and increase of myocardial stiffness. At least in a murine model these alterations were attenuated after the administration of a matrix metalloprotease inhibitor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The transition from a compensated to a decompensated stage of left ventricular hypertrophy (LVH) is accompanied by functional and structural changes. Among the latter, downregulations of cytoskeletal and sarcolemma-associated proteins have been previously described [1–3]. Based on these data, it was proposed that proteolysis of dystrophin may contribute to heart failure in the context of certain dilated cardiomyopathies [4]. Townsend et al. demonstrated in isolated myocytes that dystrophin loss modified calcium permeability of sarcolemma and increased diastolic stiffness [5]. Additionally, transgenic mdx mouse models expressed a non-functional dystrophin and presented with structural defects and increased left ventricular filling pressure. Reports of dystrophin expression levels in models of left ventricular pressure overload are scarce. In this regard, Han et al. observed dystrophin degradation in mice with severe LVH, ventricular dilation, systolic dysfunction, and pulmonary congestion [6]. Changes in myocardial stiffness were not evaluated. Recently, we demonstrated that dystrophin was degraded by matrix metalloprotease type 2 (MMP-2). This effect was inhibited by the administration of doxycycline, an MMP inhibitor that induces inhibition of MMP proteins by binding to metal ions including Ca2+ and Zn2+ [7]. Given these previous findings, it was relevant to evaluate in an LVH model without ventricular dilation whether doxycycline treatment prevented dystrophin breakdown, and consequently improved left ventricular dysfunction.
Transverse aortic constriction (TAC) models in mice have been shown to develop LVH with structural and functional characteristics similar to those observed in human subjects with aortic stenosis (AS). However, to these authors’ knowledge, no studies have evaluated the expression of dystrophin and its association with myocardial stiffness in humans with this pathology. Studies monitoring symptomatic patients after aortic valve replacement suggested that surgery did not completely reverse the structural changes that occurred in the myocardium and extracellular matrix [8]. Therefore, it was relevant to determine whether pre-surgical structural alterations correlated with 1 year post-surgical patient outcomes.
We hypothesized that dystrophin was degraded in a model of LVH and that this was positively associated with myocardial stiffness. Since MMPs functionally degrade dystrophin, treatment with doxycycline may have prevented dystrophin breakdown and attenuated increased myocardial stiffness. The first objective was to determine whether doxycycline (Doxy) prevents myocardial stiffness alterations and changes in dystrophin expression due to TAC induced LVH in mice. A second objective was to evaluate ventricular function recovery 1 year after valve replacement in human subjects, and correlate those findings to pre-surgical left ventricular end-diastolic wall stress (LVEDWS) and dystrophin levels.
Materials and methods
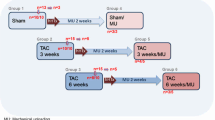
LVH in mice with aortic constriction
All protocols concerning animal use were approved by the Committee of Animal Care at the Faculty of Medicine, University of Buenos Aires (Res: 959/12). Thirty-three FVB male mice (19–24 g) were used. They were anesthetized with ketamine (65 mg/kg), xylazine (13 mg/kg), and acepromazine (2 mg/kg). All the animals received a post-surgical dose of cefazolin (50 mg/kg) and tramadol (2.5 mg/kg). The transverse thoracic aorta between the innominate artery and left common carotid artery was constricted using a 28-gage needle and a 7–0 nylon suture with the aid of a dissecting microscope and under anesthesia. After removal of the needle, the aortas remained constricted.
Animals were stratified into three groups: (1) The sham group (n = 11), in which a lateral left thoracotomy was performed, but without performing TAC. (2) The LVH group (n = 14), in which the sham group experimental protocol was repeated, but in this case TAC was performed. (3) The LVH + doxycycline group (n = 15), in which the LVH group experimental protocol was repeated, but subjects were treated with doxycycline (100 mg/kg) immediately following surgery for a period of 3 weeks. In all experimental groups, the animals were monitored for 3 weeks post-surgery.
Echocardiography
Transthoracic echocardiography was performed using an Acuson Sequoia C 512 ultrasound system with a 14-MHz linear transducer. Echocardiographic experiments were performed under light anesthesia (287.5 mg/kg of 2.5% filtered 2,2,2-tribromoethanol; Sigma–Aldrich). The two-dimensional parasternal short-axis imaging plane was used to obtain M-mode tracings at the level of the papillary muscles. LV internal dimensions and LV wall thickness (LVWT) were determined at systole and diastole using leading-edge methods and guidelines of the American Society of Echocardiography [9]. End-diastolic measurements were taken at the maximal LV diastolic dimension, and end systole was defined as the time of the most anterior systolic excursion of the posterior wall. Measurements were taken from three consecutive beats for each subject. Ejection fraction (EF) and shortening fraction (SF) were calculated and used as ejective indexes of systolic function. EF was estimated from LV dimensions by the cubed method as follows: EF (%) = [(LVEDD3−LVESD3)/LVEDD3] × 100, where LVEDD is LV end-diastolic diameter and LVESD is LV end-systolic diameter. The isovolumic relaxation time (IVRT) was measured from the Doppler-echo study.
Hemodynamics
A high-fidelity catheter (1.4-Fr Millar catheter SPR-839, Millar Instruments) was inserted into the right carotid artery and then advanced into the LV to measure LV systolic pressure (LVSP) and LV end-diastolic pressure (LVEDP). To measure the pressure gradient across the aortic constriction, the catheter was retracted to the ascending aorta, and a second catheter was inserted to the abdominal aorta through the right femoral artery. Pressures were measured simultaneously. LV end-diastolic wall stress (LVEDWS) was calculated as follows:
LVEDWS = 1.36 × (LVEDP × LVEDD)/2 LVEDWT)].
LVEDWT: Left ventricular end-diastolic wall thickness.
The LVEDWS/LVEDD ratio (g/cm3) was assessed to measure diastolic stiffness. The Tau (τ, msec), an isovolumic relaxation index, was calculated from the left ventricular pressure curve.
Isolation of mitochondria
Left ventricle mitochondria were obtained by differential centrifugation. The entire procedure was carried out at 2–4 °C. Left ventricles were excised, washed, weighed, and minced in ice-cold sodium chloride-Tris-EDTA (STE) buffer containing 250 mM sucrose, 5 mM Tris–HCl, and 2 mM EGTA, pH 7.4. A brief digestion was performed in STE medium supplemented with 0.5% (w/v) fatty acid-free BSA, 5 mM MgCl2, 1 mM ATP, and 2.5 U/mL type XXIV bacterial proteinase. After 4 min of incubation on ice, the samples were added to STE, and tissues were homogenized by eight passes with a Potter-Elvehjem glass-Teflon homogenizer, and then centrifuged at 8000×g for 10 min. The resulting pellet was re-suspended in ice-cold STE buffer and centrifuged at 700×g for 10 min. The supernatant was collected and mitochondria were precipitated from it by two 10 min centrifugations at 8000×g. Mitochondria were then re-suspended at approximately 10 mg/mL in STE buffer, which yielded 1.5–2.2 mg of cardiac mitochondrial proteins per murine subject. Mitochondrial membranes were isolated by subjecting the mitochondrial preparation to three freeze/thaw cycles, with subsequent homogenization by passage through a 29-gage hypodermic needle. Final protein concentrations were measured with the Folin’s reagent using BSA as a standard.
Mitochondrial oxygen consumption
Mitochondrial O2 uptake (MitoMVO2) was determined polarographically with a Clark-type electrode (Oxytherm, Hansatech Instruments Ltd, Norfolk, England) in a 1.0 mL chamber at 30 °C in an air-saturated reaction medium ([O2] = 220 μM). Mitochondria were suspended, at 0.2–0.3 mg protein/mL, in a respiration buffer consisting of 120 mM KCl, 5 mM KH2PO4, 1 mM EGTA, 20 mM HEPES, and 1 mg/mL fatty acid-free BSA, pH 7.40. The mitochondrial-rich preparation was added to initiate mitochondrial respiration. After reaching equilibrium, state 4 respiration rates were determined using 2 mM malate and 5 mM glutamate or 4 mM succinate as substrates of mitochondrial complexes I or II, respectively; state 3 respiration rate was established by addition of 0.5 mM ADP. Respiration was expressed in ng at O/min mg protein, and respiratory control was calculated as the ratio between state 3 and state 4 respiration rates. Oligomycin (0.5 µM) and carbonyl cyanide m-chlorophenylhydrazone (m-CPPP, 0.5 µM) were used to set state 4 and state 3, respectively.
LVH in patients with aortic stenosis
Twenty-four patients were prospectively recruited to participate in the study. Eighteen of those recruited had been diagnosed with AS and LVH (age 55 ± 3 years), while six (age 54 ± 3 years) with neither LVH nor coronary disease were recruited as controls. All participants provided informed consent for participation per the guidelines of the Declaration of Helsinki and subsequent to approval by the ethical committee of Hospital Universitario Austral.
Patients diagnosed with aortic valve stenosis were stratified based on normal LVEDWS (n = 9): <30 g/cm2; or high LVEDWS (n = 9): >30 g/cm2. All patients underwent echocardiography and cardiac catheterization studies before valve replacement surgery. Left ventricular systolic functions were evaluated by measuring ejection fractions (EF) and shortening fractions (SF). LV systolic functions were further assessed by measuring global longitudinal myocardial peak strains (GLPS) from standard two-dimensional images captured prior to and 1 year post-surgeries. Diastolic functions were assessed by calculating indices derived from LV pressure curves and echocardiography-Doppler data. LVEDWS were calculated using cylindrical models. LVEDWS/LVEDD ratios were assessed as myocardial stiffness indices. Isovolumic relaxations were evaluated using isovolumic relaxation time (IVRT) measurements.
Western blot
Biopsy samples from left ventricular anterior walls were obtained during surgery from human participants (n = 6) and at sacrifice in murine models (n = 4, per group). The samples were immediately frozen in liquid nitrogen and later homogenized in pH 7.6 buffer consisting of Tris (1.2 mM), NaCl (0.36 mM), SDS 0.1%, Triton 1%, DTT (0.2 mM), and a cocktail of protease and phosphatase inhibitors (Bio-Rad). Once homogenized, the samples were centrifuged at 26,156×g (4 °C) for 20 min. Protein concentrations were determined using Bradford assays (Bio-Rad). Samples were separated by SDS–PAGE gel electrophoresis in 5 and 18% gels and transferred to polyvinylidene fluoride (PVDF) membranes. Afterwards, the membranes were incubated with anti-dystrophin (1:1000) (MANDYS 8, Sigma) and anti-phospholamban (1:1000, Badrilla) and anti-phospho-phospholamban (Ser16) (1:1000, Badrilla) antibodies. All western blots were developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). The relative levels of dystrophin were quantified by densitometry with the Image Gauge 4.0 (Fujifilm) program and normalized to readouts from the control group. Equivalent protein loadings were confirmed using an anti-GAPDH (1:1000, Cell Signaling Technology) antibody.
Statistical analyses
Data were expressed as mean ± standard error of the mean (SEM). Comparisons between the groups were performed by variance analysis (one-way ANOVA) followed by calculation of the students’ t distribution, adjusted for multiple comparisons using the Bonferroni correction. Thus, statistical significance was not reached unless the corresponding P value was lower than 0.05/k, where k represents the number of comparisons.
Results
Murine aortic constriction models
Weights of the murine subjects, hearts, and LVH of each group are given in Table 1. Three weeks after surgery, LVW/BW and TL/BW ratios measured in both the LVH and LVH + Doxy groups significantly increased above ratios observed in the Sham group (P < 0.05 vs. Sham). No significant differences were found in heart rate, LVEDD, and LVESD between the three groups (Table 2). However, the LVEDWT was increased in the LVH and LVH + Doxy groups (P < 0.05 vs. Sham, each). Aortic-left ventricular pressure gradients were assessed to determine aortic constriction rates. LVH (88.28 ± 9.21 mmHg) and LVH + Doxy (90.67 ± 8.53 mmHg) groups both were significantly more constricted than controls (P < 0.05).
Ejection and shortening fraction measurements are given in Table 2. There were no significant differences between the groups for either of these indices. These data suggest that left ventricle pump function was preserved in the 3-week LVH group. However, contractile state (as determined by: +dP/dtmáx/LVEDWS) was significantly decreased in the LVH group compared with the Sham group. Interestingly, doxycycline treatment rescued contractile alterations observed in the LVH group.
Left ventricular diastolic function was evaluated by measuring two parameters: isovolumic relaxation and myocardial stiffness. Isovolumic relaxation was evaluated through the time constant of pressure decay (τ, msec) and the isovolumic relaxation time (IVRT, Fig. 1a–b). The τ constant was significantly increased from 6.01 ± 1.21 msec in the Sham group to 13 ± 1.61 msec (P < 0.05) in the LVH group and 12.64 ± 1.76 msec in the LVH + Doxy group. Similarly, IVRT rates significantly increased in the LVH and LVH + Doxy groups to 16.64 ± 0.65 and 16.89 ± 0.53 msec, respectively (P < 0.05 vs. Sham).

a Isovolumic relaxation was prolonged in murine models with LVH. b This alteration was not prevented with doxycycline treatment. c Total and phosphorylated (Ser16) phospholamban (PLB) expression was measured. d PLB phosphorylation was decreased in the LVH group, which was not reversed with doxycycline. *P < 0.05 versus Sham. IVRT Isovolumic relaxation time
Semi-quantitative measurements of total and phosphorylated phospholamban were obtained with western blotting techniques (Fig. 1c). Total expression of phospholamban in the LVH and LVH + Doxy was significantly elevated compared with the Sham group. However, the ratio of the phosphorylated (Ser16) phospholamban to total phospholamban protein expression decreased in the LVH and LVH + Doxy groups compared with controls (Fig. 1d, P < 0.05).
To evaluate ventricular stiffness, LVEDWS/LVEDD ratios were measured in murine models. LVEDWS/LVEDD values increased in animals with LVH (3.86 ± 0.57 g/cm3) compared with controls (1.07 ± 0.18 g/cm3; Fig. 2a, P < 0.05). Alterations in myocardial stiffness were not observed in the doxycycline-exposed group. Additionally, dystrophin protein abundance was significantly lower in the LVH group compared with controls (Fig. 2b). Alterations in dystrophin were not observed in the group exposed to 3-week doxycycline treatment. A linear correlation between dystrophin level and myocardial stiffness was observed (Fig. 2c). Finally, oxygen consumption was significantly decreased during respiration states 3 and 4 in cardiac mitochondria collected from the LVH group (P < 0.05 vs. Sham). Alterations in mitochondrial respiration were not observed in the doxycycline-treated group (Fig. 3).

a LVEDWS/LVEDD significantly increased in murine models with LVH, which was prevented by doxycycline. b The expression of dystrophin was measured. There was a significant degradation of dystrophin in the LVH group, which was completely prevented by doxycycline. c A linear correlation between the dystrophin level and the myocardial stiffness was calculated. *P < 0.05 versus Sham

Mitochondrial oxygen consumption rates in states 3 and 4 of mitochondrial respiration were measured. A significant decrease in oxygen consumption was observed in the LVH group in both states 3 and 4. Treatment with doxycycline completely reversed respiration differences. *P < 0.05 versus Sham
Prospective aortic stenosis patient data
Epidemiological data collected from consented subjects are given in Table 3. The average age of AS patients was significantly higher than the average age of the control population.
AS subjects presented with ventricular masses and aortic/ventricular pressure gradients that were elevated when compared with controls. There were no significant differences in EF and SF between the three groups (Table 4). However, +dP/dtmax/LVEDWS ratios were significantly reduced in the high-LVEDWS group when compared with both the controls as well as the normal-LVEDWS group, suggesting that contractile states were impaired in these subjects.
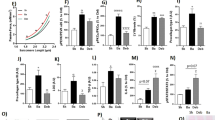
Patients with normal LVEDWS exhibited modest increases in IVRT values (NS; Fig. 4a). However, subjects with high LVEDWS experienced striking increases; the index changed from a mean of 84.01 ± 1.70 msec in the control group to 141.86 ± 21.82 msec (P < 0.05) in the high-LVEDWS group. The LVEDWS/LVEDD ratio, an index of myocardial stiffness, was 0.77 ± 0.03 g/cm3 in the control group and 0.73 ± 0.04 g/cm3 in the normal-LVEDWS group (NS), while it was increased in the high-LVEDWS group (1.11 ± 0.11 g/cm3; P < 0.05; Fig. 4b).

a Patients with normal LVEDWS experienced modestly increased IVRT values. Patients with high LVEDWS experienced significant IVRT increases compared with controls. b LVEDWS/LVEDD ratios significantly increased in the high-LVEDWS group. c Dystrophin expression was decreased in patients with high LVEDWS, and expression levels correlated with LVEDWS/LVEDD ratios. *P < 0.05 versus control group
Dystrophin expression was significantly decreased in patients with high LVEDWS (Fig. 4c). Importantly, dystrophin expression in both aortic stenosis groups correlated with LVEDWS/LVEDD ratios (Fig. 4d), suggesting that the dystrophin breakdown was related to increased myocardial stiffness. Finally, systolic function at 1 year post-surgery, demonstrated a significant recovery of the GLPS in subjects who exhibited normal LVEDWS prior to surgery (Fig. 5). However, the index remained depressed in patients with high LVEDWS prior to surgery.

Evaluation of GLPS at 1 year post-surgery demonstrated significant recovery in patients who had normal LVEDWS prior to surgery. Alternatively, patients who had high LVEDWS prior to surgery had GLPS that remained depressed at 1 year post-surgery. *P < 0.05 versus Pre
Discussion
In the present study, we observed, in both murine models and human subjects with LVH secondary to pressure overload, a significant dystrophin breakdown that was associated with systolic and diastolic dysfunctions. These dysfunctions were characterized by prolonged isovolumic relaxation times and increases in myocardial stiffness. Importantly, in the LVH murine model, doxycycline treatment attenuated increases in myocardial stiffness and dystrophin breakdowns. Also, a reduction in mitochondrial oxygen consumption was observed in the LVH murine model, suggesting an alteration in oxidative phosphorylation that may have led to increased reactive oxygen species (ROS) production.
In previous studies, the beneficial effects of doxycycline in LVH or myocardial infarction models have been described [10, 11]. However, those previous studies did not evaluate either diastolic function or mechanisms responsible for protective effects.
Interestingly, results presented here indicate that doxycycline treatments prevented contractile state alterations. Previously, Wang et al. demonstrated decreased inotropism in isolated ventricular trabeculae models in which hearts were over-expressing MMP-2 [12]. MMP-2s targeted several different proteins (troponin I, type 1 myosin light chain), potentially explaining the observed contractile dysfunctions [13, 14]. Together, these findings suggested that MMP inhibition by doxycycline treatment could improve systolic dysfunction caused by LVH.
On the other hand, in human subjects with AS, LVH, and preserve EF, we identified a subgroup with high LVEDWS. These patients presented with increased myocardial stiffness and cardiac structural changes evidenced by significant dystrophin breakdowns. In this patient subgroup, as in mice, the expression of dystrophin correlated with myocardial stiffness. The novelty of this finding was that the relationship between dystrophin expression and myocardial stiffness had only been described with in vitro studies of myocytes and in transgenic mouse models (mdx mice) [5]. The present study additionally demonstrated, in both experimental models with LVH and human subjects with high LVEDWS, that dystrophin breakdown correlated with increased myocardial stiffness. The functional and structural changes observed here were clinically relevant as they could have modified post-surgical outcomes. Indeed, ventricular function was evaluated 1 year post-surgery and a recovery of longitudinal peak strain was observed, but not in subjects that presented with high LVEDWS and dystrophin degradation prior to valve replacement.
Elevations in myocardial stiffness have been a well-described segment of LVH evolution and have been linked to the extracellular matrix [15], particularly with modifications in composition or volume of interstitial collagen [16, 17]. In the present study, we used a mouse LVH model that did not exhibit ventricular dilation or evidence of heart failure. Previously, a significant increase in interstitial collagen was not observed in these LVH models until 4 weeks of LVH evolution [18]. Alternatively, the observed increases in myocardial stiffness may have resulted from calcium transient alterations [19]. Supporting this, previous work has shown in isolated cardiac myocytes that dystrophin deficiency increased stiffness and subsequent ventricular filling pressures in intact hearts [5]. Loss of dystrophin caused sarcolemmals instability and permeability, as well as downstream cytosolic calcium and myocardial stiffness. We previously demonstrated that doxycycline treatment prevented dystrophin degradation through inhibition of matrix metalloproteinase type 2 (MMP-2) activities [7]. MMP-2 was activated by oxidative stress. In the current study, ROS production was not directly evaluated. However, reductions observed in MitoMVO2 strongly suggested that increased mitochondrial ROS occurred in LVH murine models [20]. Doxycycline prevented both dystrophin degradations and alterations in MitoMVO2. This is in accordance with the previous results that demonstrated decreased MitoMVO2 in transgenic mouse models that overexpressed MMP-2 [21]. Thus, it was possible that mitochondrial ROS production in hearts with LVH induced MMP-2 activation, which led to dystrophin degradation and increased myocardial stiffness.
On the other hand, we observed a prolonged isovolumic relaxation time both in mice and patients with LVH. This diastolic alteration is associated with a decreased ratio of the phosphorylated (Ser16) phospholamban to total phospholamban protein expression in the LVH groups compared with controls.
A first limitation of the present study was that MMP activities were not measured by zymography. However, to demonstrate the inhibition effect of doxycycline dose on MMP-2 activity, we [7] and others authors [22] added doxycycline to the zymography incubation buffer. A second limitation is that the patients with LVH have higher BMI compared to control group. However, we did not detect significant differences between patients of LVH groups.
In summary, we described here that mouse models and human subjects with LVH due to pressure overloads, but without ventricular dilation, presented systolic and diastolic dysfunctions. Diastolic dysfunctions were characterized by prolonged ventricular relaxation times and increases in myocardial stiffness. Doxycycline pre-treatment attenuated LVH-induced elevations in myocardial stiffness as well as dystrophin degradations. Thus, results of the present study are pathophysiologically important as they identify a role for dystrophin as a regulator of myocardial stiffness in LVH. Furthermore, recovery of ventricular function 1 year post-surgery solely in patients with normal LVEDWS and dystrophin level is a clinically relevant finding. Future studies should attempt to confirm these findings in larger sample populations with prolonged post-surgical follow-up times.
References
Hein S, Kostin S, Heling A, Maeno Y, Schaper J (2000) The role of the cytoskeleton in heart failure. Cardiovasc Res 45:273–278. doi:10.1016/S0008-6363(99)00268-0
Toyo-Oka T, Kawada T, Nakata J, Xie H, Urabe M, Masui F, Ebisawa T, Tezuka A, Iwasawa K, Nakajima T, Uehara Y, Kumagai H, Kostin S, Schaper J, Nakazawa M, Ozawa K (2004) Translocation and cleavage of myocardial dystrophin as a common pathway to advanced heart failure: a scheme for the progression of cardiac dysfunction. Proc Natl Acad Sci USA 101:7381–7385. doi:10.1073/pnas.0401944101
Kawada T, Masui F, Tezuka A, Ebisawa T, Kumagai H, Nakazawa M, Toyo-Oka T (2005) A novel scheme of dystrophin disruption for the progression of advanced heart failure. Biochim Biophys 1751:73–81. doi:10.1016/j.bbapap.2005.01.001
Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN, Metzger JM (2010) Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Investig 120:1140–1150. doi:10.1172/JCI41329
Townsend D, Yasuda S, McNally E, Metzger JM (2011) Distinct pathophysiological mechanisms of cardiomyopathy in hearts lacking dystrophin or the sarcoglycan complex. FASEB J 25:3106–3114. doi:10.1096/fj.10-178913
Han F, Lu YM, Hasegawa H, Kanai H, Hachimura E, Shirasaki Y, Fukunaga K (2010) Inhibition of dystrophin breakdown and endothelial nitric-oxide synthase uncoupling accounts for cytoprotection by 3-[2-[4-(3-chloro-2-methylphenyl)-1-piperazinyl]ethyl]-5,6-dimethoxy-1-(4-imidazolylmethyl)-1H-indazole dihydrochloride 3.5 hydrate (DY-9760e) in left ventricular hypertrophied mice. J Pharmacol Exp Ther 332:421–428. doi:10.1124/jpet.109.161646
Buchholz B, Perez V, Siachoque N, Miksztowicz V, Berg G, Rodríguez M, Donato M, Gelpi RJ (2014) Dystrophin proteolysis: a potential target for MMP-2 and its prevention by ischemic preconditioning. Am J Physiol Heart Circ Physiol 307:88–96. doi:10.1152/ajpheart.00242.2013
Yarbrough WM, Mukherjee R, Ikonomidis JS, Zile MR, Spinale FG (2012) Myocardial remodeling with aortic stenosis and after aortic valve replacement: mechanisms and future prognostic implications. J Thorac Cardiovasc Surg 143:656–664. doi:10.1016/j.jtcvs.2011.04.044
Sahn DJ, DeMaria A, Kisslo J, Weyman A (1978) Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation 58:1072–1083. doi:10.1161/01.CIR.58.6.1072
Errami M, Galindo CL, Tassa AT, Dimaio JM, Hill JA, Garner HR (2008) Doxycycline attenuates isoproterenol and transverse aortic banding-induced cardiac hypertrophy in mice. J Pharmacol Exp Ther 324:1196–1203. doi:10.1124/jpet.107.133975
Villarreal FJ, Griffin M, Omens J, Dillmann W, Nguyen J, Covell J (2003) Early short-term treatment with doxycycline modulates postinfarction left ventricular remodeling. Circulation 108:1487–1492. doi:10.1161/01.CIR.0000089090.05757.34
Wang G, Bergman M, Nguyen A, Turcato S, Swigart P, Rodrigo M, Simpson PC, Karliner JS, Lovett DH, Baker AJ (2006) Cardiac transgenic matrix metalloproteinase-2 expression directly induces impaired contractility. Cardiovasc Res 69:688–696. doi:10.1016/j.cardiores.2005.08.023
Rork TH, Hadzimichalis NM, Kappil MA, Merrill GF (2006) Acetaminophen attenuates peroxynitrite-activated matrix metalloproteinase-2-mediated troponin I cleavage in the isolated guinea pig myocardium. J Mol Cell Cardiol 40:553–561. doi:10.1016/j.yjmcc.2006.01.010
Sawicki G, Leon H, Sawicka J, Sariahmetoglu M, Schulze CJ, Scott PG, Szczesna-Cordary D, Schulz R (2005) Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury: a new intracellular target for matrix metalloproteinase-2. Circulation 112:544–552. doi:10.1161/CIRCULATIONAHA.104.531616
Jalil JE, Doering CW, Janicki JS, Pick R, Shroff SG, Weber KT (1989) Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ Res 64:1041–1050. doi:10.1161/01.RES.64.6.1041
Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, Kinugawa S, Tsutsui H (2006) Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension 47:711–717. doi:10.1161/01.HYP.0000208840.30778.00
Conrad CH, Brooks WW, Hayes JA, Sen S, Robinson KG, Bing OH (1995) Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation 91:161–170. doi:10.1161/01.CIR.91.1.161
Gelpi RJ, Gao S, Zhai P, Yan L, Hong C, Danridge LM, Ge H, Maejima Y, Donato M, Yokota M, Molkentin JD, Vatner DE, Vatner SF, Sadoshima J (2009) Genetic inhibition of calcineurin induces diastolic dysfunction in mice with chronic pressure overload. Am J Physiol Heart Circ Physiol 297:1814–1819. doi:10.1152/ajpheart.00449.2009
Stuyvers BD, Miura M, ter Keurs HE (2000) Ca (2+) dependence of passive properties of cardiac sarcomeres. Adv Exp Med Biol 481:353–366
Akhmedov AT, Rybin V, Marín-García J (2015) Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail Rev 20:227–249. doi:10.1007/s10741-014-9457-4
Zhou LY, Liu JP, Wang K, Gao J, Ding SL, Jiao JQ, Li PF (2013) Mitochondrial function in cardiac hypertrophy. Int J Cardiol 167:1118–1125. doi:10.1016/j.ijcard.2012.09.082
Cheung PY, Sawicki G, Wozniak M, Wang W, Radomski MW, Schulz R (2000) Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation 101:1833–1839 doi:10.1161/01.CIR.101.15.1833
Acknowledgements
We thank Dr. Liliana Grinfeld for assistance in recruiting patients in the control group and for additional invaluable support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
none declared.
Additional information
Martín Donato, Bruno Buchholz, Laura Valdez, Tamara Zaobornyj, Alberto Boveris, and Ricardo J. Gelpi are members of the National Council of Scientific and Technological Research (CONICET).
Rights and permissions
About this article

Cite this article
Donato, M., Buchholz, B., Morales, C. et al. Loss of dystrophin is associated with increased myocardial stiffness in a model of left ventricular hypertrophy. Mol Cell Biochem 432, 169–178 (2017). https://doi.org/10.1007/s11010-017-3007-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-3007-z