
Abstract
Malignant glioma is the most common and aggressive form of brain tumor with poor prognosis of survival. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a promising anticancer agent but is insufficient of inducing apoptosis in some types of gliomas. In this study, we showed that the small-molecule Mcl-1 inhibitor UMI-77 sensitized glioma cells to TRAIL treatment, as evidenced by cell viability assay, Annexin V staining and JC-1 staining. Combination of UMI-77 and TRAIL in glioma cells led to the activation of caspase-8 and Bid, cleavage of caspase-3 and poly-ADP ribose polymerase (PARP), accumulation of tBid in the mitochondria and release of cytochrome c into the cytosol. UMI-77 alone or in combination with TRAIL untethered pro-apoptotic Bcl-2 proteins Bim and Bak from the sequestration of Mcl-1 and promoted the conformational activation of Bak. Small hairpin RNA (shRNA) of Bid attenuated the cleavage of caspase-8, Bid, caspase-3 and PARP, and reduced the cytotoxicity of UMI-77 plus TRAIL as compared with control shRNA cells, indicating this synergy entails the crosstalk between extrinsic and intrinsic apoptotic signaling. Taken together, UMI-77 enhances TRAIL-induced apoptosis by unsequestering Bim and Bak, which provides a novel therapeutic strategy for the treatment of gliomas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) belongs to the tumor necrosis family (TNF) of cytokines that are involved in inflammation and immune surveillance. TRAIL is a promising anticancer agent due to its capacity to induce apoptosis in a broad range of cancer cells while sparing normal cells. TRAIL activates the death receptor (extrinsic) signaling upon binding to its functional receptors DR4 (TRAIL-R1) and DR5 (TRAIL-R2), resulting in receptor trimerization and recruitment of Fas-associated death domain (FADD), caspase-8 and caspase-10 to form the death-inducing signaling complex (DISC) [1–3]. DISC assembly induces caspase-8 cleavage and activation. In the so-called ‘type I’ cells, caspase-8 activation is sufficient to activate caspase-3 and trigger apoptosis [4], while in ‘type II’ cells, the caspase-8-dependent cleavage of Bid and its downstream mitochondrial (intrinsic) apoptotic signaling are essential to fully activate caspase-3. Truncated Bid (tBid) translocates to the mitochondria and activates Bax and Bak, which leads to mitochondrial outer membrane permeabilization (MOMP), release of mitochondrial cytochrome c, smac/DIABLO and AIF into the cytosol and consequently activation of caspase-3 and apoptosis [5–8].
Unfortunately, innate or acquired TRAIL resistance of cancer cells limits TRAIL-based therapy. Overexpression and TRAIL-stimulated transcriptional activation of pro-survival proteins of Bcl-2 family are parts of the reason [9]. Bcl-2, Bcl-xL and Mcl-1 bind to pro-apoptotic Bax and Bak, preventing their conformational changes and activation. The pro-apoptotic BH3-only proteins bind to specific pro-survival proteins and prevent the latter to bind Bax and Bak or directly activate them [10]. Several small molecules that mimic BH3-only proteins have been utilized to promote TRAIL treatment. For instance, the Bad-like BH3 mimetic ABT-737 that targets Bcl-2 and Bcl-xL potentiates TRAIL-induced apoptosis by abolishing sequestration of Bim and Bak from Bcl-2 or Bcl-xL in pancreatic cancer cells and by promoting tBid mitochondrial accumulation in glioma cells [11, 12]. Although BH3 mimetics targeting Mcl-1 were developed previously and their anticancer effect was proven when used alone or in combination with the Bcl-2/Bcl-xL inhibitor ABT-263, their synergistic apoptosis-inducing effect with TRAIL has not been tested [13, 14]. To date, strategies targeting Mcl-1 for TRAIL sensitization are mostly indirect, either by inhibiting its transcription or increasing its protein instability to reduce its protein level [15, 16]. In this study, we sought to determine whether the Mcl-1-specific inhibitor UMI-77 sensitizes human glioma cells to TRAIL-induced apoptosis. Our results showed that UMI-77 sensitized glioma cells to TRAIL by releasing sequestered Bak and Bim from Mcl-1.
Materials and methods
Cell culture
Human glioma cell lines U87, U251 and A172 and the cervical cancer cell line HeLa were obtained from the Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). U87 and U251 cells were cultured in DMEM plus 10% fetal bovine serum (FBS), 1% non-essential amino acid and 1% sodium pyruvate (Life technologies, Grand Island, USA). A172 cells were cultured in DMEM plus 10% FBS. HeLa cells were cultured in MEM plus 10% FBS. All the cells were maintained under standard cell culture conditions at 37 °C and 5% CO2.
Reagents
We used antibodies specific for Bax, Bak, Bcl-2, Bid, Bim, caspase-3, caspase-8, Mcl-1, poly-ADP ribose polymerase (PARP), horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies (Cell Signaling Technology, Beverly, USA), Bcl-xL (Santa Cruz Technology, Santa Cruz, USA), Bax (6A7), Mcl-1 (for immunoprecipitation) (BD Biosciences, San Jose, USA), Bak, NT (Merck Millipore, Darmstadt, Germany) and GAPDH (Kangchen, Shanghai, China). Annexin V Apoptosis Detection Kit APC (eBioscience, San Diego, USA), Propidium iodide (PI, Sigma-Aldrich), protease inhibitor cocktail, RNase A (Thermo Scientific, Waltham, USA), MTT, phosphatase inhibitor (Sangon, Shanghai, China), cell lysis buffer for Western and IP, cytosolic and mitochondrial protein extraction kit, mitochondrial membrane potential assay kit with JC-1, protein A agarose, protein G agarose, PMSF, RIPA, (Beyotime, Nantong, China), UMI-77 (Selleck, Shanghai, China) and human recombinant TRAIL (ProSpec, Ness Ziona, Israel) were used in this study.
Lentivirus-mediated Gene transduction
Short hairpin RNAs (shRNAs) targeting human Mcl-1 (CCCTAGCAACCTAGCCAGAAA), Bid (CCGTGATGTCTTTCACACA) and the scrambled (control) shRNA (TTCTCCGAACGTGTCACGT) were inserted into the lentiviral vector pLKD-CMV-GFP-U6-shRNA (Neuron Biotech, Shanghai, China). Human non-degradable phospho-defective Mcl-1 (T92A) mutant was generated by PCR amplification and inserted into the lentiviral vector pLOV-EF1a-eGFP (Neuron Biotech, Shanghai, China). Insertion at indicated site was confirmed by DNA sequencing. Lentiviruses encoding various shRNA plasmids and expression plasmids were produced as previously described [17].
Western blot
After collection, cells were lysed in RIPA supplemented with PMSF, phosphatase inhibitor and protease inhibitor cocktail. Western blot was carried out as previously described [17].
MTT assay
To evaluate the half maximal inhibitory concentration (IC50) of UMI-77, U87, U251 and A172 cells were plated in 96-well plates (3000 cells/well) and treated with indicated doses of UMI-77 for 48 h. To evaluate the synergistic cytotoxicity of UMI-77 and TRAIL, U87 and A172 cells were plated in 96-well plates (3000 cells/well) and treated with UMI-77 (0, 4, 8 and 12 μM) for 24 h and additional with TRAIL (0, 10, 30 and 100 ng/ml) for 24 h. Following treatment, the cell viability was determined by MTT assay as previously described [17].
Flow cytometry
To measure apoptosis, cells were stained with Annexin V according to the manufacturer’s protocol of the Annexin V Apoptosis Detection Kit APC, or with PI as previously described [17]. To measure the mitochondrial membrane potential, cells were stained with JC-1 according to the manufacturer’s protocol of mitochondrial membrane potential assay kit with JC-1. After staining, cells were analyzed by BD LSR II flow cytometer. Data were analyzed by FlowJo software.
Cell fractionation assay
The cytosolic and mitochondrial protein of cells was fractionated using cytosolic and mitochondrial protein extraction kit according to the manufacturer’s protocol.
Immunoprecipitation
After treatment, cells were harvested and subjected to immunoprecipitation as previously described [18].
Statistical analysis
Data analysis was performed using Microsoft Excel and OriginPro 8 software. Values represent the mean ± standard error of the mean (SEM). The statistical significance of the differences between experimental groups was determined with Student’s t test.
Results
UMI-77 synergizes with TRAIL to induce apoptosis in glioma cells
To determine whether Mcl-1 plays a key role in TRAIL resistance of glioma cells, we knocked down Mcl-1 in three human glioma cell lines, U87, U251 and A172, followed by treating them with different concentrations of recombinant human TRAIL (30 ng/ml for U87, 100 ng/ml for U251 and 10 ng/ml for A172). Neither Mcl-1 knockdown (shMcl-1) nor TRAIL had significant effect on apoptosis, whereas shMcl-1 plus TRAIL potently induced apoptosis in U87 and A172 cells (Fig. 1a, b). Combination of shMcl-1 and TRAIL had little effect on U251 cells probably because both Bcl-2 and Bcl-xL were increased in U251 cells (Fig. 1c), which compensated for loss of Mcl-1 to retain anti-apoptotic effects. Although A172 cells are innately sensitive to TRAIL [19], they were used in the following experiments to test the synergistic effect of Mcl-1 loss and TRAIL, as cell apoptosis induced by relatively low dose of TRAIL (10 ng/ml) is enhanced by Mcl-1 knockdown (Fig. 1a, b).

Mcl-1 knockdown augments TRAIL-induced apoptosis in U87 and A172 glioma cells. a and b U87, U251 and A172 cells infected with lentiviruses encoding either control shRNA or Mcl-1 shRNA (shMcl-1) were treated with TRAIL (30 ng/ml for U87, 100 ng/ml for U251 and 10 ng/ml for A172) for 24 h and stained by Annexin V-APC antibody. The percentage of apoptotic cells was measured by flow cytometry. Representative data were shown in (a) and averaged values were shown in (b) (n = 3, mean ± SEM, *p < 0.05). c Levels of proteins in control shRNA and shMcl-1-expressing U87, U251 and A172 cells were analyzed by Western blot. GAPDH was used as a loading control
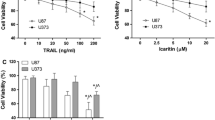
UMI-77 was identified as a selective binding inhibitor of Mcl-1 that induced apoptosis in pancreatic cancer cells by blocking the interaction of Mcl-1 with Bax and Bak [14]. Here we evaluated the cytotoxic effect of UMI-77 in glioma cells. IC50 of UMI-77 after a 48 h treatment was 3.72 μM in HeLa cells that are strictly dependent on Mcl-1 for survival [20], and were relatively higher in U87, U251 and A172 cells, showing functional redundancy of anti-apoptotic Bcl-2 proteins in these cells (Fig. 2a). When combined with TRAIL, 8 μM or higher concentration of UMI-77 lowered viabilities of UMI-77-resistant U87 and A172 cells (Fig. 2b). Combination of 8 μM (lowest effective concentration) UMI-77 and TRAIL resulted in higher levels of cleaved caspase-8, Bid, caspase-3 and PARP, the accumulation of tBid in the mitochondria and the release of cytochrome c into cytosol in U87 and A172 cells, suggesting the onset of MOMP and caspase-dependent apoptosis (Fig. 2c, d).

UMI-77 sensitizes U87 and A172 glioma cells to TRAIL-induced apoptosis. a To calculate the half maximal inhibitory concentration (IC50) values, U87, U251, A172 and HeLa cells were treated with indicated concentration of UMI-77 for 48 h and subjected to MTT assay. b U87 and A172 cells were treated with indicated concentration of UMI-77 for 24 h and further treated with indicated concentration of TRAIL for 24 h. The cell viability was analyzed by MTT assay. c U87 and A172 cells were treated with UMI-77 (8 μM) for 24 h and further treated with TRAIL (30 ng/ml) for indicated period of time. Levels of apoptosis-associated proteins were analyzed by Western blot. GAPDH was used as a loading control. d U87 and A172 cells were treated with UMI-77 (8 μM) for 24 h and further treated with TRAIL (30 ng/ml for U87 and 10 ng/ml for A172) for 6 h. Levels of proteins in the cytosolic and mitochondrial fractions of cells were analyzed by Western blot. β-tubulin and COX4 were used as the loading control of cytosolic and mitochondrial fraction, respectively
The synergistic apoptotic effect of UMI-77 and TRAIL is dependent on Mcl-1
To investigate whether the effect of UMI-77 in synergizing apoptosis with TRAIL is dependent on Mcl-1, degradation-resistant Mcl-1 (T92A) mutant was moderately overexpressed using EF1a promoter in U87 and A172 cells. Mcl-1 overexpression compromised apoptosis as well as cleavage of caspase-8, Bid, caspase-3 and PARP in U87 and A172 cells treated with UMI-77 and TRAIL (Fig. 3a–c). JC-1 staining showed that loss of mitochondrial outer membrane potential upon combination treatment was partly inhibited by Mcl-1 overexpression in U87 and A172 cells (Fig. 4a, b). Moreover, 8 μM UMI-77 did not further improve TRAIL-induced apoptosis in the Mcl-1 knockdown U87 cells because the synergistic effect of shMcl-1 and TRAIL was already greater than that of UMI-77 and TRAIL (Fig. 5). These results suggest that glioma cell apoptosis induced by UMI-77 and TRAIL combination is dependent on Mcl-1.

Mcl-1 overexpression attenuates apoptosis induced by UMI-77 plus TRAIL. a and b U87 and A172 cells infected with lentiviruses encoding either an empty vector (control OE) or Mcl-1 (T92A) were treated with UMI-77 (8 μM) for 24 h and further treated with TRAIL (30 ng/ml for U87 and 10 ng/ml for A172) for 24 h. After Annexin V-APC staining, the percentage of apoptotic cells was analyzed by flow cytometry. Representative data were shown in (a). Values from three independent experiments were averaged and shown in (b) (n = 3, mean ± SEM, *p < 0.05). c Levels of apoptosis-associated proteins of U87 and A172 cells treated as in (a) were analyzed by Western blot. GAPDH was used as a loading control

Mcl-1 overexpression attenuates loss of mitochondrial membrane potential (MMP) induced by UMI-77 plus TRAIL. a and b Control OE and Mcl-1 (T92A)-expressing U87 and A172 cells were treated with UMI-77 (8 μM) for 24h and further treated with TRAIL (30 ng/ml for U87 and 10 ng/ml for A172) for 6 h. Cells were incubated with JC-1 and subjected to flow cytometry for measuring JC-1 aggregates and monomers. Representative data were shown in (a) and averaged values of the percentage of cells with MMP loss were shown in (b) (n = 3, mean ± SEM, *p < 0.05)

UMI-77 fails to improve apoptosis induced by TRAIL and Mcl-1 knockdown in U87 cells. Control shRNA and shMcl-1-expressing U87 cells were treated with UMI-77 (8 μM) for 24 h and further treated with TRAIL (30 ng/ml) for 24 h. Levels of apoptosis-associated proteins were analyzed by Western blot. GAPDH was used as a loading control
UMI-77 unsequesters Bim and Bak from Mcl-1 and induces Bak activation, alone or in combination with TRAIL
To study the mechanism behind the combination-induced apoptosis, we first performed immunoprecipitation to evaluate the binding of pro-apoptotic Bcl-2 proteins with Mcl-1 in UMI-77-treated U87 cells. At less as 8 μM, UMI-77 untethered Bim and Bak, two pro-apoptotic Bcl-2 proteins that normally bound to Mcl-1 [21], from the sequestration of Mcl-1 in a dose-dependent manner (Fig. 6a). Bax was hardly detected in Mcl-1 precipitants, but there was a tendency that UMI-77 also displaced Bax from Mcl-1. Next, we examined the binding of these pro-apoptotic proteins to Mcl-1 in U87 cells treated with UMI-77 and TRAIL. UMI-77 promoted dissociation of Bak and Bim from Mcl-1 and Bcl-xL/Bax interaction without affecting Bcl-2/Bim and Bcl-xL/Bak interaction. On the other hand, TRAIL enhanced tBid/Bcl-xL interaction and the binding of Mcl-1 to Bim and Bak (Fig. 6b). UMI-77 and TRAIL combination reversed TRAIL-induced binding of Mcl-1 to Bim and Bak, enhanced TRAIL-induced Bid truncation, and inhibited UMI-77-induced Bcl-xL/Bax interaction. In addition, UMI-77 and TRAIL, respectively, upregulated conformational active forms of Bak and Bax, whereas their combination activated Bax and Bak simultaneously (Fig. 6b). These results suggest that UMI-77 frees Bak and Bim from Mcl-1 and activates Bak with or without TRAIL.

UMI-77 displaces Bim and Bak from the sequestration by Mcl-1. a U87 cells treated with indicated concentration of UMI-77 for 12 h and Mcl-1 was immunoprecipitated by the corresponding antibody. Levels of proteins in precipitants and total cell lysates were analyzed by Western blot. b U87 cells were treated with UMI-77 (8 μM) for 24 h and further treated with TRAIL (30 ng/ml) for 6 h. Bcl-xL, Bim, Mcl-1 and conformational active Bax and Bak were immunoprecipitated by corresponding antibodies. Levels of proteins in precipitants (left panel) and lysates (right panel) were analyzed by Western blot. GAPDH was used as a loading control of lysates
Bid is important for apoptosis induced by UMI-77 and TRAIL combination
Given that UMI-77 promoted crosstalk between the mitochondrial apoptotic signaling and the death receptor signaling which is induced by TRAIL, we investigated the role of Bid, the linker of these two signaling, in mediating such crosstalk in U87 cells. Bid knockdown suppressed cleavage of caspase-8, caspase-3 and PARP and cell death induced by UMI-77 plus TRAIL (Fig. 7a, b). These results underscore the importance of Bid in the cell death induced by UMI-77 and TRAIL combination.

Bid is required for UMI-77 and TRAIL-induced apoptosis. Control shRNA and shBid-expressing U87 cells were treated with UMI-77 (8 μM) for 24 h and further treated with TRAIL (30 ng/ml) for 24 h. a Levels of apoptosis-associated proteins were analyzed by Western blot. GAPDH was used as the loading control. b The percentage of apoptotic cells in sub-G1 phase was analyzed by flow cytometry (n = 3, mean ± SEM, *p < 0.05)
Discussion
TRAIL receptor agonists (TRA) have been demonstrated as well tolerated agents but with disappointing anticancer efficacy in clinical trials [22]. Combinations of TRA with other targeted therapies are of interest to override TRA resistance in cancer cells. In this study, we demonstrate that Mcl-1 is a key determinant of TRAIL resistance in glioma cells. The Mcl-1 inhibitor UMI-77 synergizes with TRAIL to induce apoptosis in glioma cells through releasing Bak and Bim from Mcl-1.
The importance of Mcl-1 in TRAIL resistance galvanizes the development of combination therapies of TRAIL with inhibition of Mcl-1 mostly by lowering its expression, including mitotic interfering agents induced proteasome degradation of Mcl-1, CDK9 inhibitors induced transcriptional elongation blockage and consequent decrease of Mcl-1 mRNA and NF-κB or STAT3 inhibitors induced transcription inhibition of Mcl-1 [15, 16, 21, 23, 24]. However, study combining TRAIL with direct inhibition of Mcl-1 is lacking. UMI-77 is a Noxa-like BH3 mimetic that specifically binds and inhibits Mcl-1 among anti-apoptotic proteins of Bcl-2 family. It shows promising anticancer efficacy in a panel of pancreatic cancer cell lines [14]. Furthermore, UMI-77 sensitizes pancreatic cancer cells to radiation with no harm to normal cells [25]. However, IC50s of UMI-77 in some of the cell lines are quite higher than those in others, possibly due to functional redundancy of pro-survival Bcl-2 proteins. Similarly, IC50s in glioma cells are higher than those in HeLa cells and UMI-77-sensitive pancreatic cells. Raising UMI-77 doses to fully inhibit Mcl-1 could improve curative effects but may cause adverse effects like cardiac failure as Mcl-1 is required for normal mitochondrial physiology [26, 27]. Given that as high as 60 mg/kg UMI-77 inhibited the progression of UMI-77-sensitive pancreatic cells in a xenograft mouse model whereas 80 mg/kg UMI-77 is lethal to mice [14], UMI-77 monotherapy is unsuitable for in vivo inhibition of Mcl-1-independent tumors. Partial inhibition of Mcl-1 within the therapeutic window by UMI-77 plus TRAIL, however, is feasible for both TRAIL- and UMI-77-resistant cancer cells. Apart from displacing of Bak from Mcl-1, which is demonstrated in our study and by others [14], UMI-77 promotes the dissociation of BimEL from Mcl-1, resulting in the activation of Bak. However, UMI-77 also promotes the binding of Bcl-xL to Bax, which potentially dampens Bax activation. On the other hand, TRAIL increases Bim/Mcl-1 complex, probably because tBid preferentially binds and disrupts the binding of Bcl-xL rather than Mcl-1 to Bim upon TRAIL stimulation [28]. Bim is a BH3-only protein that potentiates apoptosis either by neutralizing anti-apoptotic Bcl-2 proteins, activating Bax and Bak directly or transforming Bcl-2 into a pro-apoptotic protein [29, 30]. Hence, simultaneous inhibition of Bcl-xL and Mcl-1 by TRAIL and UMI-77 respectively freed Bax, Bak and Bim to the greatest extent that surmounts the apoptosis threshold.
In conclusion, the Mcl-1 selective inhibitor UMI-77 exerts on-target apoptotic-inducing effect in combination with TRAIL, which will potentially benefit patients with gliomas. Future explorations are required to investigate whether UMI-77 plus TRAIL or other targeted therapies may be extended to the treatment of other malignancies.
Abbreviations
- TRAIL :
-
Tumor necrosis factor-related apoptosis-inducing ligand
- Mcl-1 :
-
Myeloid cell leukemia-1
- PARP :
-
Poly-ADP ribose polymerase
- shRNA :
-
Small hairpin RNA
- Bcl :
-
B cell lymphoma
- Bid :
-
BH3-interacting domain death agonist
- BH :
-
Bcl-2 homology
- tBid :
-
Truncated Bid
- TNF :
-
Tumor necrosis factor
- FADD :
-
Fas-associated death domain
- DISC :
-
Death-inducing signaling complex
- MOMP :
-
Mitochondrial outer membrane permeabilization
References
Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, Timour MS, Gerhart MJ, Schooley KA, Smith CA, Goodwin RG, Rauch CT (1997) TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. Embo j 16:5386–5397. doi:10.1093/emboj/16.17.5386
Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM (1997) The receptor for the cytotoxic ligand TRAIL. Science 276:111–113
Wilson NS, Dixit V, Ashkenazi A (2009) Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol 10:348–355. doi:10.1038/ni.1714
Gonzalvez F, Ashkenazi A (2010) New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 29:4752–4765. doi:10.1038/onc.2010.221
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87:99–163. doi:10.1152/physrev.00013.2006
Lemke J, von Karstedt S, Zinngrebe J, Walczak H (2014) Getting TRAIL back on track for cancer therapy. Cell Death Differ 21:1350–1364. doi:10.1038/cdd.2014.81
Yamada H, Tada-Oikawa S, Uchida A, Kawanishi S (1999) TRAIL causes cleavage of bid by caspase-8 and loss of mitochondrial membrane potential resulting in apoptosis in BJAB cells. Biochem Biophys Res Commun 265:130–133. doi:10.1006/bbrc.1999.1641
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481–490
Son JK, Varadarajan S, Bratton SB (2010) TRAIL-activated stress kinases suppress apoptosis through transcriptional upregulation of MCL-1. Cell Death Differ 17:1288–1301. doi:10.1038/cdd.2010.9
Weber K, Harper N, Schwabe J and Cohen GM (2013) BIM-mediated membrane insertion of the BAK pore domain is an essential requirement for apoptosis. Cell Rep 5:409–420. doi:10.1016/j.celrep.2013.09.010
Huang S, Sinicrope FA (2008) BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res 68:2944–2951. doi:10.1158/0008-5472.CAN-07-2508
Cristofanon S, Fulda S (2012) ABT-737 promotes tBid mitochondrial accumulation to enhance TRAIL-induced apoptosis in glioblastoma cells. Cell Death Dis 3:e432. doi:10.1038/cddis.2012.163
Xiao Y, Nimmer P, Sheppard GS, Bruncko M, Hessler P, Lu X, Roberts-Rapp L, Pappano WN, Elmore SW, Souers AJ, Leverson JD, Phillips DC (2015) MCL-1 Is a key determinant of breast cancer cell survival: validation of MCL-1 dependency utilizing a highly selective small molecule inhibitor. Mol Cancer Ther 14:1837–1847. doi:10.1158/1535-7163.MCT-14-0928
Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS, Gulappa T, Cierpicki T, Owens S, Zhang T, Sun D, Stuckey JA, Mohammad RM, Nikolovska-Coleska Z (2014) A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther 13:565–575. doi:10.1158/1535-7163.MCT-12-0767
Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W, Jin Z, Liu YY, Dicker DT, Chiao PJ, Flaherty KT, Smith CD, El-Deiry WS (2007) Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 12:66–80. doi:10.1016/j.ccr.2007.05.006
16.Lemke J, von Karstedt S, Abd El Hay M, Conti A, Arce F, Montinaro A, Papenfuss K, El-Bahrawy MA, Walczak H (2014) Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1. Cell Death Differ 21:491–502. doi:10.1038/cdd.2013.179
Zhu Z, Li K, Xu D, Liu Y, Tang H, Xie Q, Xie L, Liu J, Wang H, Gong Y, Hu Z, Zheng J (2013) ZFX regulates glioma cell proliferation and survival in vitro and in vivo. J Neurooncol 112:17–25. doi:10.1007/s11060-012-1032-z
Zhu Z, Liu Y, Li K, Liu J, Wang H, Sun B, Xiong Z, Jiang H, Zheng J, Hu Z (2014) Protein tyrosine phosphatase receptor U (PTPRU) is required for glioma growth and motility. Carcinogenesis 35:1901–1910. doi:10.1093/carcin/bgu123
van Roosmalen IA, Reis CR, Setroikromo R, Yuvaraj S, Joseph JV, Tepper PG, Kruyt FA and Quax WJ (2014) The ER stress inducer DMC enhances TRAIL-induced apoptosis in glioblastoma. Springerplus 3:495. doi:10.1186/2193-1801-3-495
Eichhorn JM, Alford SE, Sakurikar N, Chambers TC (2014) Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp Cell Res 322:415–424. doi:10.1016/j.yexcr.2014.02.010
Lee DH, Sung KS, Bartlett DL, Kwon YT, Lee YJ (2015) HSP90 inhibitor NVP-AUY922 enhances TRAIL-induced apoptosis by suppressing the JAK2-STAT3-Mcl-1 signal transduction pathway in colorectal cancer cells. Cell Signal 27:293–305. doi:10.1016/j.cellsig.2014.11.013
den Hollander MW, Gietema JA, de Jong S, Walenkamp AM, Reyners AK, Oldenhuis CN, de Vries EG (2013) Translating TRAIL-receptor targeting agents to the clinic. Cancer Lett 332:194–201. doi:10.1016/j.canlet.2012.04.007
Sanchez-Perez T, Ortiz-Ferron G, Lopez-Rivas A (2010) Mitotic arrest and JNK-induced proteasomal degradation of FLIP and Mcl-1 are key events in the sensitization of breast tumor cells to TRAIL by antimicrotubule agents. Cell Death Differ 17:883–894. doi:10.1038/cdd.2009.176
Murphy AC, Weyhenmeyer B, Noonan J, Kilbride SM, Schimansky S, Loh KP, Kogel D, Letai AG, Prehn JH, Murphy BM (2014) Modulation of Mcl-1 sensitizes glioblastoma to TRAIL-induced apoptosis. Apoptosis 19:629–642. doi:10.1007/s10495-013-0935-2
Wei D, Zhang Q, Schreiber JS, Parsels LA, Abulwerdi FA, Kausar T, Lawrence TS, Sun Y, Nikolovska-Coleska Z, Morgan MA (2015) Targeting mcl-1 for radiosensitization of pancreatic cancers. Transl Oncol 8:47–54. doi:10.1016/j.tranon.2014.12.004
Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, Opferman JT (2013) Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev 27:1351–1364. doi:10.1101/gad.215855.113
Perciavalle RM, Opferman JT (2013) Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol 23:22–29. doi:10.1016/j.tcb.2012.08.011
Meng XW, Lee SH, Dai H, Loegering D, Yu C, Flatten K, Schneider P, Dai NT, Kumar SK, Smith BD, Karp JE, Adjei AA, Kaufmann SH (2007) Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43-9006)-induced TRAIL sensitization. J Biol Chem 282:29831–29846. doi:10.1074/jbc.M706110200
Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O’Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD, Bouillet P (2009) The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol 186:355–362. doi:10.1083/jcb.200905153
Zhao L, He F, Liu H, Zhu Y, Tian W, Gao P, He H, Yue W, Lei X, Ni B, Wang X, Jin H, Hao X, Lin J, Chen Q (2012) Natural diterpenoid compound elevates expression of Bim protein, which interacts with antiapoptotic protein Bcl-2, converting it to proapoptotic Bax-like molecule. J Biol Chem 287:1054–1065. doi:10.1074/jbc.M111.264481
Acknowledgements
Financial supports from the Natural Science Foundation of Shanghai (Grant Number 15ZR1409700) are acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Liu, JW., Zhu, ZC., Li, K. et al. UMI-77 primes glioma cells for TRAIL-induced apoptosis by unsequestering Bim and Bak from Mcl-1. Mol Cell Biochem 432, 55–65 (2017). https://doi.org/10.1007/s11010-017-2997-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-2997-x