
Abstract
In a recent study, we highlighted the importance of cationic charge and arginine residues for the neuroprotective properties of poly-arginine and arginine-rich peptides. In this study, using cortical neuronal cultures and an in vitro glutamic acid excitotoxicity model, we examined the neuroprotective efficacy of different modifications to the poly-arginine-9 peptide (R9). We compared an unmodified R9 peptide with R9 peptides containing the following modifications: (i) C-terminal amidation (R9-NH2); (ii) N-terminal acetylation (Ac-R9); (iii) C-terminal amidation with N-terminal acetylation (Ac-R9-NH2); and (iv) C-terminal amidation with d-amino acids (R9D-NH2). The three C-terminal amidated peptides (R9-NH2, Ac-R9-NH2, and R9D-NH2) displayed neuroprotective effects greater than the unmodified R9 peptide, while the N-terminal acetylated peptide (Ac-R9) had reduced efficacy. Using the R9-NH2 peptide, neuroprotection could be induced with a 10 min peptide pre-treatment, 1–6 h before glutamic acid insult, or when added to neuronal cultures up to 45 min post-insult. In addition, all peptides were capable of reducing glutamic acid-mediated neuronal intracellular calcium influx, in a manner that reflected their neuroprotective efficacy. This study further highlights the neuroprotective properties of poly-arginine peptides and provides insight into peptide modifications that affect efficacy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neuronal cell death as a result of glutamic acid receptor-mediated excitotoxicity is associated with several neurological disorders such as stroke, global cerebral ischaemia, traumatic brain injury, and perinatal hypoxic ischaemic encephalopathy [1–4]. Despite the neurological impact of glutamic acid excitotoxicity and its downstream damaging processes, there is still no clinically relevant neuroprotective pharmacotherapy for neurological disorders. Therefore, there is an urgent need to identify clinically effective and widely applicable therapies aimed towards reducing neuronal cell death following stroke and other clinical conditions associated with excitotoxicity. Recently, it has been demonstrated that poly-arginine (e.g. R9) and arginine-rich peptides (e.g. TAT, penetratin), which belong to a class of peptides with cell-penetrating properties are neuroprotective in vitro and in animal models of stroke [5–10]. For example, we have previously demonstrated that poly-arginine peptides (R8–R18) and some other arginine-rich peptides are neuroprotective in vitro in neurons exposed to glutamic acid excitotoxicity and oxygen glucose deprivation, and in the case of R9, R12, and R18, after permanent middle cerebral artery occlusion in the rat [6, 8, 9]. Moreover, it was demonstrated that peptide neuroprotective efficacy increases with the increase in arginine content, and that peptide cationic charge and arginine residues are critical for neuroprotection [6].
In terms of neuroprotective action, we have also demonstrated that poly-arginine peptides have the capacity to reduce glutamic acid-induced neuronal calcium influx and are neuroprotective with a single treatment 1–4 h before, immediately before, during, or after glutamic acid or OGD exposure [6]. The neuroprotective effect induced by the poly-arginine peptides could be blocked by incubation with the highly negatively charged molecule heparin, or by neutralising peptide charge with the addition of negatively charged glutamic acid amino residues (E) fused to the poly-arginine peptide (E9/R9 peptide) [6]. The ability of heparin and glutamic acid residues to inhibit poly-arginine peptide neuroprotection is evidence that peptide binding to negatively charged cell surface receptors or membrane structures, such as heparan sulphate proteoglycans (HSPGs), sialic acid residues on glycosphingolipids, or phosphate head groups in membrane phospholipids which are known to stimulate peptide endocytosis/uptake [11–13], is a critical factor for neuroprotection; however, the peptide–heparin interaction is also likely to nullify other potential neuroprotective actions (e.g. perturbation of cationic ion channels).
In order for poly-arginine peptides to be developed into neuroprotective therapeutic agents, it will be essential to determine if specific chemical modifications can further enhance peptide efficacy and/or peptide stability and to obtain additional information regarding potential mechanism of action. Therefore, in the present study, we examined the influence of R9 peptide modifications on neuroprotective properties, peptide stability, and calcium influx inhibition when applied to cortical neuronal cultures exposed to glutamic acid excitotoxicity. Comparative studies utilised an unmodified R9 peptide and R9 peptides with C-terminal amidation, N-terminal acetylation, C-terminal amidation/N-terminal acetylation, and C-terminal amidation/d-amino acid modifications.
Materials and methods
Neuronal cultures
Establishment of rat primary cortical cultures in Neurobasal (NB)/2% B27 supplement (B27) (Life Technologies, Australia) using cortical tissue obtained directly from E18-day embryos was as previously described [5]; however, cultures were established from cortical tissue stored in Hibernate-E (Life Technologies)/2% B27 for 2–7 days at 5 °C. Neurons were seeded into 96-well-sized glass wells (7 mm diameter, Grace, Australia), 96-well plastic plates (Nunc, Thermo Fisher Scientific, Australia), or 96-well plastic strip-plates (Costar, Sigma-Aldrich, Australia) and maintained in a CO2 incubator (5% CO2, 95% air balance, 98% humidity) at 37 °C until use on day in vitro 10–14. Under these conditions, cultures routinely consist of >97% neurons and 1–3% astrocytes. Glass and strip wells were used for calcium kinetic studies.
Peptides used in study
Peptides used in this study are provided in Table 1. Peptides were synthesised by China Peptides (Shanghai, China) and purified by high-performance liquid chromatography. The R9D-NH2 peptide was synthesised in the protease-resistant d-isoform, using d-amino acids. All the peptides were resuspended in water (Baxter, Australia; 500 µM) and assessed in a concentration range anywhere from 0.1 to 20 µM, dependent upon experimental application.
Glutamic acid excitotoxicity model and peptide treatments
Peptides were added to culture wells 15 min prior to glutamic acid (l-glutamic acid; Sigma-Aldrich) exposure by removing media and adding 50 µL of NB/2% B27 containing the specific peptide. To induce excitotoxicity, 50 µL of NB/2% B27 containing glutamic acid (200 µM; final concentration 100 µM) was added to the culture wells and incubated at 37 °C in the CO2 incubator for 5 min. After the 5 min exposure, media was replaced with 100 µL of NB/2% B27, and cultures were incubated for a further 24 h at 37 °C in the CO2 incubator. Untreated controls with or without glutamic acid treatment underwent the same incubation steps and media additions. Note: In this model, neurons are exposed to peptide for 15 min prior to and during (half peptide concentration) the 5-min glutamic acid insult.
For extended pre-glutamic acid exposure experiments, neurons were exposed to peptide for a 10-min period only 1, 2, 3, 4, 5, or 6 h before glutamic acid exposure. This was performed by removing media from wells and adding 50 µL of NB/2% B27 containing peptide at the relevant pre-treatment time point. After 10-min at 37 °C in the CO2 incubator, media were removed and replaced with 100 µL of NB/2% B27. Subsequently, media were removed from wells and replaced with 100 µL of NB/2% B27 containing glutamic acid (100 µM). After 5-min glutamic acid exposure, neuronal cultures were treated as described above. Untreated controls with or without glutamic acid treatment underwent the same incubation steps and media as the 1-h pre-glutamic acid-treated cultures.
Post-glutamic acid exposure was performed by replacing media in wells with 100 µL of NB/2% B27 containing glutamic acid (100 µM). After 5-min glutamic acid exposure, media were replaced with 100 µL of NB/2% B27 with (0-min post-insult time-point) or without peptide and placed at 37 °C in the CO2 incubator. In addition, for the 15, 30, and 45-min post-glutamic acid exposure time points media were replaced with 100 µL of NB/2% B27 with peptide. Untreated controls with or without glutamic acid treatment underwent the same incubation steps and media additions as the 0-min post-glutamic time point-treated cultures.
Peptide cytotoxicity studies
Peptide cytotoxicity was assessed by exposing neuronal cultures to different peptide concentrations for 24 h. Twenty-four exposures consisted of removing media from culture wells and adding 100 µL of NB/2% B27 containing peptide at 1, 2, 5, or 10 µM continuously for 24 h at 37 °C in the CO2 incubator.
Peptide stability studies
Conditioned culture media were harvested from 12-day old neuronal cultures and clarified by centrifugation. The conditioned media were used to make-up peptide stock concentrations (0.5, 1, 2, and 5 µM) for each of the R9 modified peptides and solutions incubated at 37 °C in the CO2 incubator for 24 h. Following the 24 h incubation, 50 µL of conditioned culture media containing peptide were added to neuronal culture wells and wells treated as described above for the glutamic acid excitotoxicity model. Controls were treated with conditioned media that had undergone the same incubation steps as media containing peptide.
Heparin experiments
Peptides were pre-incubated with heparin (20 IU/mL; Pfizer, Australia) in NB/2% B27 for 5 min at room temperature before addition to culture wells (50 µL) for 15 min at 37 °C in the CO2 incubator. After the incubation period, media in wells were removed and replaced with 100 µL of NB/2% B27 containing glutamic acid (100 µM) and subsequently treated as described above. For all the experiments, non-heparin-treated peptide controls with glutamic acid treatment underwent the same incubation steps and media additions.
Intracellular calcium kinetics
Intracellular calcium influx as monitored in neuronal culture wells (glass wells) as previously described [6]. The aim of these experiments was to determine the relative change in intracellular calcium before and after glutamic acid exposure. Briefly, cells were loaded with the fluorescent calcium-ion indicator Fura-2 AM (5 µM; Sigma-Aldrich, Australia) in 50 µL NB/2% B27, 0.1% pluronic F-127, for 20 min at 37 °C (5% CO2). Fura-2 AM solution was removed from wells, replaced with 50 µL NB/2% B27 containing peptide or glutamate receptor blockers (MK801/CNQX; 5 µM/5 µM) and incubated for 10 min at 37 °C (5% CO2). Control cultures received 50 µL of NB/2% B27 only. After the 10 min incubation period, media in wells were replaced with 50 µL of balanced salt solution (mM: 116 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgSO4, 1 NaH2PO4; pH 7.2) and wells were transferred to a spectrophotometer (BMG Labtec, CLARIOstar, Australia) while maintaining temperature at 37 °C. Fifty microliters of NB/2% B27 containing glutamic acid (200 µM; final concentration 100 µM) was added to wells, and every 5 s, starting 30 s before and for 2 min after glutamic acid addition, spectrophotometer measurements (excitation: 355 nm/emission 495 nm) were recorded. Experiments were performed in triplicate.
Neuronal cell viability
Neuronal viability was quantitatively measured by MTS (3-(4,5,dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfo-phenyl-2H-tetrazolium salt) assay (Promega, Australia), which involved adding 20 µL of MTS solution to neuronal culture wells and incubating for 1–3 h at 37 °C (5% CO2), as per manufacturers guidelines. In the glutamic acid model, the MTS absorbance (490 nm) data were converted to reflect proportional cell viability relative to both the no insult control (Cont.) and glutamic acid-treated control (Glut.), with the untreated control taken as 100% viability. After glutamic acid exposure, cell death in these control wells typically ranges from 2 to 5%, based on light microscopy.
Statistical analysis
Viability data were analysed by ANOVA, followed by post hoc Fisher’s protected least significant difference test, with P < 0.05 values considered statistically significant. All data are represented as mean ± standard error of the mean (SEM). Experiments were repeated independently at least two times.
Results
Peptide toxicity
We first examined the cytotoxic effects of different concentrations (1, 2, 5, and 10 µM) of the modified R9 peptides in neuronal cultures following a 24-h exposure duration (Fig. 1). Following the 24-h constant exposure of peptides to neuronal cultures, the only peptide that appeared to cause any significant increase in neuronal death compared to the untreated neuronal cultures was the R9D-NH2 peptide. At the 5 and 10 µM peptide concentrations, R9D-NH2 reduced neuronal metabolism of the MTS substrate by 47 and 61%, respectively, when compared to the control. Interestingly, at least one concentration dose of all peptides increased the ability of neurons to metabolise the MTS substrate above control levels, an observation we have reported previously with R9 peptide treatment of neuronal cultures following glutamic acid excitotoxicity [4], and likely represents a direct or indirect effect of poly-arginine peptides increasing the capacity of neurons to reduce MTS to its formazan product and/or to reduce background cell death in control neuronal cultures.

Glutamic acid excitotoxicity model; assessment of R9 peptide modifications on peptide stability. Peptides incubated for 24 h in conditioned culture media before addition to neuronal cultures for 15 min prior to, and during, a 5-min glutamic acid exposure. Neuronal viability measured 24 h after glutamic acid exposure. Concentration of peptide in µM. MTS data expressed as percentage neuronal viability with no insult control taken as 100% viability (mean ± SEM n = 4; *P < 0.05, when compared to Glut). Cont no peptide no glutamic acid-treated control. Glut no peptide glutamic acid treated control
Peptide dose response studies
Next, we examined the influence of N-terminal acetylation and/or C-terminal amidation and d-isomer amino acids on the neuroprotective efficacy of the R9 peptide. The R9 peptide was used in our previous study [6] and was used as the benchmark for the other modified R9 peptides. The use of peptide end terminal acetylation/amidation and d-isoform amino acids is known to increase peptide resistance to proteases [14]. Additionally, acetylation decreases peptide net charge by 1 (−1) due to the removal of a positively charged N-terminal amino group, and amidation increases peptide net charge by 1 (+1) by removal of a negatively charged C-terminal carboxyl group [14].
We observed that the C-terminal amidated peptides (Ac-R9-NH2 and R9-NH2; net charge +9 and +10, respectively), and the d-amino acid amidated peptide (R9D-NH2; net charge +10) displayed a similar and an increased level of neuroprotective efficacy, respectively, when compared to the unmodified R9 parent peptide (net charge +9) (Fig. 2; see 2 µM results). Conversely, the N-terminal acetylated peptide Ac-R9 (net charge +8) displayed significantly reduced neuroprotective efficacy when compared to the unmodified R9 peptide; for experiments shown in Fig. 2, the increased level of neuronal survival obtained with the Ac-R9 peptide only reached statistical significance at the 5 µM concentration.

Glutamic acid excitotoxicity model; R9 peptide modification dose response experiment. Peptides present in neuronal cultures for 15 min prior to, and during, 5-min glutamic acid exposure. Neuronal viability measured 24 h after glutamic acid exposure. Concentration of peptide in µM. MTS data expressed as percentage neuronal viability with no insult control taken as 100% viability (mean ± SEM n = 4; *P < 0.05, when compared to Glut). Cont no peptide no glutamic acid-treated control. Glut no peptide glutamic acid-treated control
Effect of delayed peptide pre-treatment
In previous experiments, peptides were present in neuronal cultures 10 min before and during (half concentration) the 5-min glutamic acid insult. In this study, we tested the neuroprotective efficacy of the R9-NH2 peptides when added to neuronal cultures at 1, 2, 3, 4, 5, or 6-h time interval (10 min treatment; 5 or 10 µM) before glutamic acid insult. We observed that for R9-NH2, peptide pre-treatment of neuronal cultures was neuroprotective at the 1–6 h time intervals and that the effect was time and dose dependent (Fig. 3).

Glutamic acid excitotoxicity model; assessment of R9 peptide treatment before glutamic acid exposure. R9-NH2 peptide present in neuronal cultures for 10 min only at 1–6 h before glutamic acid exposure. Neuronal viability measured 24 h after glutamic acid exposure. Concentration of peptide in µM. MTS data expressed as percentage neuronal viability with no insult control taken as 100% viability (mean ± SEM n = 4; *P < 0.05, when compared to Glut). Cont no peptide no glutamic acid-treated control. Glut no peptide glutamic acid-treated control
Effect of post-glutamate peptide treatment
We next assessed the neuroprotective efficacy of the R9-NH2 peptide when added to neuronal cultures at different time points (0, 15, 30, and 45 min) post-glutamic acid insult. We observed that R9-NH2 was neuroprotective when added after the glutamic acid insult, however, neuroprotective efficacy was significantly reduced compared to the pre-treatment effects (Fig. 4).

Glutamic acid excitotoxicity model; assessment of R9 peptide treatment after glutamic acid exposure. R9-NH2 peptide present in neuronal cultures after 5-min glutamic acid exposure at different time points (0–45 min). Neuronal viability measured 24 h after glutamic acid exposure. Time of peptide addition post-insult in minutes. MTS data expressed as percentage neuronal viability with no insult control taken as 100% viability (mean ± SEM n = 4; *P < 0.05, when compared to Glut). Cont no peptide no glutamic acid-treated control. Glut no peptide glutamic acid-treated control
Effect of heparin on peptide neuroprotection
To determine if peptide electrostatic interactions with negatively charged cell surface receptors (e.g. heparan sulphate proteoglycans and glycosphingolipids) were important for neuroprotection, as previously reported for R9D-NH2 and other poly-arginine peptides [6], we pre-incubated R9, Ac-R9, Ac-R9-NH2, R9-NH2, and R9D-NH2 with heparin (negative-charged molecule homologous with aforementioned cell surface receptors) during exposure to neuronal cultures (10 min treatment) and prior to glutamic acid insult. We observed that the presence of heparin completely eliminated peptide neuroprotective efficacy (Fig. 5). Exposure of neuronal cultures to heparin alone did not cause any neuronal toxicity (data not shown).

Glutamic acid excitotoxicity model; peptides incubated ± heparin (20 IU/mL) for 5 min at room temperature before being added to neuronal cultures for 10 min only and then removed prior to glutamic acid exposure. Neuronal viability measured 24 h after glutamic acid exposure. Concentration of peptide in µM. MTS data expressed as percentage neuronal viability with no insult control taken as 100% viability (mean ± SEM n = 4; *P < 0.05, when compared to Glut). Cont no peptide no glutamic acid-treated control. Glut no peptide glutamic acid-treated control. +Hep peptide incubated with heparin, −Hep no heparin
Peptide stability
Following a 24-h incubation at 37 °C in conditioned neuronal culture media, peptide stability was assessed by determining neuroprotective efficacy in the glutamic acid excitotoxicity model. The only peptide that retained any significant neuroprotective activity was the R9D-NH2 peptide (Fig. 6). At the 2 and 5 µM R9D-NH2 peptide concentrations, neuronal survival was 50 and 100%, respectively, while at the lower concentrations (0.5 and 1 µM), the peptide was ineffective.

Effect of prolonged peptide exposure on neuronal viability. R9 peptides present in neuronal cultures for 24 h prior to viability assessment. Concentration of peptide in µM. MTS data expressed as MTS absorbance (mean ± SEM n = 4; *P < 0.05, when compared to Cont.). Cont no peptide no glutamic acid-treated control
Intracellular calcium kinetics
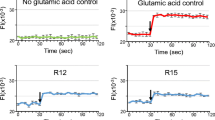
In a previous study, we showed that the R9D-NH2 peptide attenuated neuronal intracellular calcium influx immediately following glutamic acid insult [6]. Therefore, we decided to compare the dose-dependent effects of R9, Ac-R9, Ac-R9-NH2, R9-NH2, and R9D-NH2 on intracellular calcium levels in neuronal cultures following glutamic acid exposure. All peptides, to varying degrees, reduced neuronal intracellular calcium levels when administered immediately prior to (10 min treatment) glutamic acid insult (Fig. 7). Interestingly, while the R9 and Ac-R9-NH2 peptides displayed a dose-dependent effect at reducing intracellular calcium levels, the R9-NH2 and R9D-NH2 peptides reduced intracellular calcium to similar levels for all three concentrations tested (1, 2, and 5 µM). The Ac-R9 peptide only had a very modest effective at reducing neuronal calcium influx at the 2 and 5 µM concentrations.

Intracellular calcium assessment using Fura-2 AM after glutamic acid exposure in neuronal cultures. Fluorescent tracers; fluorescence intensity (FI) of neuronal cultures 30 s before and after the addition (arrow) of glutamic acid (100 µmol/L final concentration). Peptides or glutamate receptor blockers (MK801/CNQX) were added to neuronal cultures for 10 min and removed (time = 0) before glutamic acid addition. Glutamic acid control received glutamic acid exposure only. Control did not receive peptide or glutamic acid exposure. Values are mean ± SEM; n = 3
Discussion
This study extends the findings of our previous reports demonstrating the neuroprotective efficacy and calcium influx inhibitory properties of R9 and other poly-arginine peptides using in vitro glutamic acid excitotoxicity and oxygen-glucose deprivation models [5–7]. In particular, this study has made a number of novel findings that have implications for improving poly-arginine peptide neuroprotective efficacy and for gaining a better understanding of neuroprotective mechanism of action.
With respect to neuroprotection in the glutamic acid excitotoxicity model, when compared to the unmodified R9 (net charge +9) parent peptide, N-terminal amidation (Ac-R9-NH2; net charge +9, R9-NH2; net charge +10, and R9D-NH2; net charge +10) increased the neuroprotection, while N-terminal acetylation (Ac-R9; net charge +8) decreased the neuroprotection in primary neuronal cultures. The neuroprotective effects associated with N-terminal amidation were largely mirrored with the ability of the peptides to reduce neuronal calcium influx, with the R9-NH2 and R9D-NH2 peptides being the most effective across the dose range examined, and the Ac-R9 peptide being the least effective. We have previously hypothesised that one mechanism, whereby poly-arginine, reduce calcium influx associated with excitotoxicity is by inducing the internalisation cell surface ion channel receptors and transporters [7]. To this end, we have recently demonstrated that poly-arginine R12 and the TAT-fused neuroprotective peptide TAT-NR2B9c down-regulate the cell surface expression of the NMDA NR2B subunit protein in cortical neuronal cultures [15], indicating that this is likely a general property of cationic arginine-rich peptides.
The increased neuroprotective efficacy of C-terminal amidated R9 peptides could be due to increased peptide charge and/or increased peptide stability. As noted in our previous study [6], peptide net charge is an important chemical determinant with respect to neuroprotection. The influence of peptide charge on neuroprotection is further supported by the reduced efficacy of the N-terminal acetylated R9 peptide (Ac-R9), which has a net charge of +8 as compared to +9 for the unmodified R9 peptide. The reduced efficacy of Ac-R9 due to reduced charge is consistent with the decreased efficacy of R8 (net charge +8) compared to R9 reported in our earlier study [6]. Interestingly, the reduced neuroprotective effect of N-terminal acetylation (Ac-R9) can be effectively overcome and improved beyond the parent R9 peptide with N-terminal amidation (Ac-R9-NH2; net charge +9), suggesting other chemical/physical factors besides peptide net charge can also influence peptide neuroprotective function.
The d-isoform R9D-NH2 peptide did not display any significant improvement in neuroprotective efficacy compared to the l-isoform R9-NH2 peptide in the glutamic acid excitotoxicity model. This is not totally unexpected, as the neuroprotective effect induced by peptide treatment of neuronal cultures occurs within minutes [6], and therefore, peptide stability, in terms of reduced degradation, is unlikely to offer a significant advantage in the glutamic acid model. Notwithstanding this result, the stability of the R9D-NH2 peptide was confirmed following the demonstration that the peptide, unlike the other R9 peptides (R9, Ac-R9, R9-NH2, and Ac-R9-NH2), retained neuroprotective properties in the glutamic acid model following a 24-h incubation in conditioned neuronal culture media at 37 °C. The increased stability of R9D-NH2 is also the likely reason why this peptide displayed cytotoxic effects in neuronal cultures at high concentrations (5 and 10 µM) following a 24-h exposure period. With respect to in vivo efficacy and toxicity, we have already confirmed that the R9D-NH2 peptide is neuroprotective in a stroke model [6] without causing any evidence of histological damage to the kidney and liver (unpublished observation). In addition, a safety study in humans using Ac-R9D-NH2 (ALX40-4C) at doses 50, 150, 300, 400, or 500 nmol/kg administered intravenously three times per week over 4 weeks to asymptomatic HIV-positive patients was well tolerated and not associated with serious side effects [16]. However, it still remains to be determined if d-isoform peptides (e.g. R9D-NH2) due to their increased plasma half-life and resistance to proteolytic degradation are more efficacious than their l-isoform counterparts (e.g. R9-NH2) following stroke. Studies in our laboratory are planned to directly compare l- and d-isoform poly-arginine peptides in a stroke model to address this question.
In line with our previous poly-arginine peptide studies [6], R9 peptides were neuroprotective with extended pre- and post-glutamic acid treatments of neuronal cultures and displayed a time and concentration-dependent neuroprotective effect. Possible explanations for the time and concentration-dependent pre-treatment effects could be related to the extended time required to degrade higher concentrations of the peptide and/or the time to fully reverse the peptide-induced dose-dependent neuroprotective effects (e.g. recovery of NMDA receptor cell surface levels [15]).
The findings in this study provide additional data to support a neuroprotective mechanism involving poly-arginine peptides inducing internalisation of cell surface ion receptors and channels, especially with respect to reducing neuronal intracellular calcium influx immediately following glutamic acid exposure and in light of our study demonstrating the ability of R12 to down-regulate cell surface levels of NR2B [15]. In addition, in the current and/or our previous study, we have demonstrated that co-incubation of poly-arginine peptides R9, R12, and R15 with heparin, a highly negatively charged molecule, during peptide treatment of neurons completely attenuated neuroprotective efficacy following glutamic acid excitotoxicity. The inhibitory effect of heparin on peptide neuroprotection is consistent with an electrostatic interaction occurring with negatively charged cell surface structures such as heparan sulphate proteoglycans, chondroitin sulphate proteoglycans, glycosphingolipids, and phosphate head groups in phospholipids binding positively charged poly-arginine peptides and inducing endocytic uptake or membrane transduction of the peptide [7, 17]. Furthermore, inhibiting access of the peptide to the cell cytoplasm and/or the heparin–peptide interaction will also prevent the peptide from exerting any potential intracellular neuroprotective actions (e.g. mitochondrial stabilisation).
As part of the uptake process induced by poly-arginine and arginine-rich peptides, we hypothesised that cell surface receptors associated with excitotoxic calcium influx (e.g. NMDA receptor, AMPA receptor, and voltage gated calcium channels) are also internalised [7]. As mentioned above, the demonstration that the R12 peptide reduces neuronal cell surface NR2B levels [15] is supportive of this hypothesis. Also of interest is the lack of stereo-selectivity with respect to neuroprotection of the R9 peptides (i.e. l-isoform and d-isoform peptides). This is inline with a more general neuroprotective mechanism of action (e.g. glutamate receptor internalisation) for poly-arginine and arginine-rich peptides, likely mediated by their positive charge, rather than inhibition of a specific protein–protein interaction, which is likely to be sequence- and stereoselective.
While other studies have also shown that arginine-rich peptides including putative “neuroprotective peptides” fused to TAT can interfere with neuronal calcium receptor function [18–26] or cause receptor internalisation/reduced cell surface levels [20, 21, 25], other neuroprotective mechanisms are also likely. For example, it has been demonstrated that cationic arginine-rich peptides (R4, C-R7, SS-31) have the capacity to target mitochondria, whereby they can preserve ATP synthesis, reduce reactive oxygen species production, attenuate loss of the mitochondrial transmembrane potential, maintain cytochrome c integrity, and/or increase mitochondrial resistance to the toxic effects of calcium influx [10, 27–30].
Conclusion
In conclusion, we believe that poly-arginine and arginine-rich peptides due to their multiple potential beneficial biological actions represent a new class of neuroprotective molecule, which could be tailored for the treatment of a range of neurological disorders. However, additional studies are required in order to better elucidate and confirm peptide neuroprotective mechanisms and to modify peptide sequences to further improve neuroprotective efficacy.
References
Faden AI, Demediuk P, Panter SS, Vink R (1989) The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244:798–800
Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M (2002) Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298:846–850
Wu Q, Chen W, Sinha B, Tu Y, Manning S, Thomas N, Zhou S, Jiang H, Ma H, Kroessler DA, Yao J, Li Z, Inder TE, Wang X (2015) Neuroprotective agents for neonatal hypoxic-ischemic brain injury. Drug Discov Today 20:1372–1381
Hoque A, Hossain MI, Ameen SS, Ang CS, Williamson N, Ng DC, Chueh AC, Roulston C, Cheng HC (2016) A beacon of hope in stroke therapy—blockade of pathologically activated cellular events in excitotoxic neuronal death as potential neuroprotective strategies. Pharmacol Ther 160:159–179
Meloni BP, Craig AJ, Milech N, Hopkins RM, Watt PM, Knuckey NW (2014) The neuroprotective efficacy of cell-penetrating peptides TAT, penetratin, Arg-9, Pep-1 in glutamic acid, kainic acid, in vitro ischemia injury models using primary cortical neuronal cultures. Cell Mol Neurobiol 34:173–181
Meloni BP, Brookes LM, Clark VW, Cross JL, Edwards AB, Anderton RS, Hopkins RM, Hoffmann K, Knuckey NW (2015) Poly-arginine and arginine-rich peptides are neuroprotective in stroke models. JCBFM 35:993–1004
Meloni BP, Milani D, Edwards AB, Anderton RS, O’Hare Doig RL, Fitzgerald M, Palmer NT, Knuckey NW (2015) Neuroprotective peptides fused to arginine-rich cell penetrating peptides: neuroprotective mechanism likely mediated by peptide endocytic properties. Pharmacol Ther 153:36–54
Milani D, Clark VW, Cross JL, Anderton RS, Knuckey NW, Meloni BP (2016) Poly-arginine peptides reduce infarct volume in a permanent middle cerebral artery rat stroke model. BMC Neurosci 17:19–27
Milani D, Knuckey NW, Anderton RS, Cross JL, Meloni BP (2016) The R18 polyarginine peptide is more effective than the TAT-NR2B9c (NA-1) peptide when administered 60 minutes after permanent middle cerebral artery occlusion in the rat. Stroke Res Treat 2016:1–9
Marshall J, Wong KY, Rupasinghe CN, Tiwari R, Zhao X, Berberoglu ED, Sinkler C, liu J, Lee I, Parang K, Spaller MR, Hüttemann M, Goebel DJ (2015) Inhibition of N-methyl-d-aspartate-induced retinal neuronal death by polyarginine peptides is linked to the attenuation of stress-induced hyperpolarization of the inner mitochondrial membrane potential. J Biol Chem 290:22030–22048
Wallbrecher R, Verdurmen WP, Schmidt S, Bovee-Geurts PH, Broecker F, Reinhardt A, van Kuppevelt TH, Seeberger PH, Brock R (2014) The stoichiometry of peptide-heparan sulfate binding as a determinant of uptake efficiency of cell-penetrating peptides. Cell Mol Life Sci 71:2717–2729
Favretto ME, Wallbrecher R, Schmidt S, van de Putte R, Brock R (2014) Glycosaminoglycans in the cellular uptake of drug delivery vectors—bystanders or active players? J Control Release 180:81–90
Rothbard JB, Jessop TC, Wender PA (2005) Adaptive translocation: the role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv Drug Deliv Rev 57:495–504
Brugidou J, Legrand C, Mery J, Rabie A (1995) The retro-inverso form of a homeobox-derived short peptide is rapidly internalised by cultured neurones: a new basis for an efficient intracellular delivery system. Biochem Biophys Res Commun 214:685–693
McDougall G, Anderton RS, Edwards AB, Knuckey NW, Meloni BP (2016) The neuroprotective peptide poly-arginine-12 (R12) reduces cell surface levels of NMDA NR2B receptor subunit in cortical neurons; investigation into the involvement of endocytic mechanisms. J Mol Neurosci (in press)
Doranz BJ, Filion LG, Diaz-Mitoma F, Sitar DS, Sahai J, Baribaud F, Orsini MJ, Benovic JL, Cameron W, Doms RW (2001) Safe use of the CXCR4 inhibitor ALX40-4C in humans. AIDS Res Hum Retrovir 17:475–486
Vaslin A, Naegele-Tollardo S, Puyal J, Clarke PG (2011) Excitotoxicity-induced endocytosis mediates neuroprotection by TAT-peptide-linked JNK inhibitor. J Neurochem 119:1243–1252
Ferrer-Montiel AV, Merino JM, Blondelle SE, Perez-Paya E, Houghten RA, Montal M (1998) Selected peptides targeted to the NMDA receptor channel protect neurons from excitotoxic death. Nat Biotechnol 16:286–291
Planells-Cases R, Aracil A, Merino JM, Gallar J, Perez-Paya E, Belmonte C, Gonzalez-Ros JM, Ferrer-Montiel AV (2000) Arginine-rich peptides are blockers of VR-1 channels with analgesic activity. FEBS Lett 481:131–136
Fan J, Cowan CM, Zhang LYJ, Hayden MR, Raymond LA (2009) Interaction of postsynaptic density protein-95 with NMDA receptors influences excitotoxicity in the yeast artificial chromosome mouse model of Huntington’s disease. J Neurosci 29:10928–10938
Sinai L, Duffy S, Roder JC (2010) Src inhibition reduces NR2B surface expression and synaptic plasticity in the amygdala. Learn Mem 17:364–371
Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, Lew F, Chan SL, Chen Y, Lu Y (2010) DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 140:222–234
Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM, Molosh AI, You H, Hudmon A, Shekhar A, White FA, Zamponi GW, Brustovetsky N, Chen J, Khanna R (2011) Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J Biol Chem 286:37778–37792
Feldman P, Khanna R (2013) Challenging the catechism of therapeutics for chronic neuropathic pain: targeting CaV2.2 interactions with CRMP2 peptides. Neurosci lett 557:27–36
Brustovetsky T, Pellman JJ, Yang XF, Khanna R, Brustovetsky N (2014) Collapsin response mediator protein 2 (CRMP2) interacts with N-methyl-d-aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity. J Biol Chem 289:7470–7482
Garcia-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, Bladen C, Chen L, Hamid J, Pizzoccaro A, Deage M, François A, Bourinet E, Zamponi GW (2014) The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 83:1144–1158
Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO (2008) Mitochondria-penetrating peptides. Chem Biol 15:375–382
Rigobello MP, Barzon E, Marin O, Bindoli A (1995) Effect of polycation peptides on mitochondrial permeability transition. Biochem Biophys Res Commun 217:144–149
Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH (2004) Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279:34682–34690
Birk AV, Liu S, Soong YY, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto HH (2013) The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. JASN 24:1250–1261
Acknowledgements
This study in part was supported by the Department of Neurosurgery, Sir Charles Gairdner Hospital, and a University of Notre Dame Australia RIS grant, and a Neurotrauma Research Program of Western Australia grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Compliance with ethical standards
All experimental procedures in this study adhered to the guidelines approved and specified by the Animal Care Committee of the University of Western Australia, in accordance with the Policies and Guidelines of the National Health and Medical Research Council, Australia.
Conflict of interest
B.P. Meloni and N.W. Knuckey are named inventors of several patent applications regarding the use of arginine-rich peptides as neuroprotective agents. A.B. Edwards, and R.S. Anderton declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Edwards, A.B., Anderton, R.S., Knuckey, N.W. et al. Characterisation of neuroprotective efficacy of modified poly-arginine-9 (R9) peptides using a neuronal glutamic acid excitotoxicity model. Mol Cell Biochem 426, 75–85 (2017). https://doi.org/10.1007/s11010-016-2882-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2882-z