
Abstract
The carnitine/acylcarnitine transporter (CACT; SLC25A20) mediates an antiport reaction allowing entry of acyl moieties in the form of acylcarnitines into the mitochondrial matrix and exit of free carnitine. The transport function of CACT is crucial for the β-oxidation pathway. In this work, it has been found that CACT is partially acetylated in rat liver mitochondria as demonstrated by anti-acetyl-lys antibody immunostaining. Acetylation was reversed by the deacetylase Sirtuin 3 in the presence of NAD+. After treatment of the mitochondrial extract with the deacetylase, the CACT activity, assayed in proteoliposomes, increased. The half-saturation constant of the CACT was not influenced, while the V max was increased by deacetylation. Sirtuin 3 was not able to deacetylate the CACT when incubation was performed in intact mitoplasts, indicating that the acetylation sites are located in the mitochondrial matrix. Prediction on the localization of acetylated residues by bioinformatics correlates well with the experimental data. Recombinant CACT treated with acetyl-CoA was partially acetylated by non-enzymatic mechanism with a corresponding decrease of transport activity. The experimental data indicate that acetylation of CACT inhibits its transport activity, and thus may contribute to the regulation of the mitochondrial β-oxidation pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Protein lysine acetylation is a well-acknowledged post-translational modification (PTM) of a huge number of proteins in cellular regulation [1]. Enzymes, such as acetyltransferases and deacetylases, are engaged in this important PTM [2]. Investigations by mass spectrometry allowed us to identify more than 6800 acetylation sites in human proteins among some mitochondrial carriers [3–5]. Acetylation of the citrate transporter has been recently described [6]. Moreover, it was shown that acetylation of mitochondrial proteins also occurs by non-enzymatic pathway [7, 8]. Many mitochondrial proteins have been identified to be acetylated under conditions of reduced acetyl-CoA buffering [7]. Among the different metabolic implications, acetylation has been recognized as a PTM able to regulate the β-oxidation pathway. Iper-acetylation of LCAD (long-chain acyl-CoA dehydrogenase) reduces its enzymatic activity [9]. On this basis, it could be hypothesized that other components of the β-oxidation could be regulated by a similar PTM, such as the carnitine/acylcarnitine transporter (CACT), that is a putative site of regulation of the pathway [10, 11]. Acetylated peptides belonging to CACT are indeed present in raw data from proteomic analyses [12]. The functional properties of this transport protein have been defined mainly in proteoliposome experimental model reconstituted with both the native protein extracted from rat liver mitochondria and the recombinant protein obtained by overexpression in E. coli. The transporter is inserted in a right-side-out orientation into the proteoliposomal membrane with respect to the mitochondrial membrane. CACT catalyzes carnitine/acylcarnitine and carnitine/carnitine antiports and a slower substrate uniport. The carnitine/acylcarnitine antiport is involved in the shuttling of acyl units from cytosolic acyl-CoA to mitochondrial CoA. While the uniport function acts in equilibrating the cytosolic and mitochondrial carnitine concentrations, the substrate antiport occurs via a ping-pong transport mechanism. The recombinant CACT resembles all the main functional and kinetic properties of the native protein [13–15]. Site-directed mutagenesis and chemical labeling using SH reagents highlighted several structure–function relationships of the CACT [14, 16] validating the homology structural model, which was obtained by bioinformatics on the basis of the 3D structure of the ADP/ATP carrier (AAC) [17]. On the basis of this model, the CACT is constituted by a bundle of six transmembrane hydrophobic segments delimiting a water-filled cavity which is opened towards the cytosol (intermembrane space) in the c-state conformation. The residues of the substrate binding site are exposed in this cavity [13]. In the present work, CACT acetylation and structure/function relationships have been investigated.
Materials and methods
Materials
l-[Methyl-3H]carnitine from Scopus Research BV Costerweg, Sephadex G-75, egg-yolk phospholipids (l-α-phosphatidylcholine from fresh turkey egg yolk), PIPES, Triton X-100, cardiolipin, l-carnitine, protein G agarose, Sirtuin 3, nicotinamide (NAM), NAD+ from Sigma–Aldrich. Antibody anti-acetyl-lys clone 4G12 were purchased from Millipore. All other reagents were of analytical grade.
SDS-PAGE and western blot analysis
Polyacrylamide gel electrophoresis was performed in the presence of 0.1% SDS (SDS-PAGE) using the method of Laemmli [18]. Gel sizes were 8 cm × 10 cm × 0.75 mm in thickness. An acrylamide/bisacrylamide ratio of 30:0.2 was used. Proteins were transferred to nitrocellulose membrane (Schleicher and Schuell, PROTRAN BA 85 cellulose nitrate). After blotting, membranes were incubated for 30 min with a buffer containing 3% BSA or dry milk, 150 mM NaCl, 50 mM Tris–HCl pH 7.0, 0.05% Tween 20 (immunoblotting buffer). Then membranes were incubated for 1 h with in house-produced [19] anti-CACT antibody at a dilution of 1:1.000 in the presence of 0.5% BSA or overnight with mouse monoclonal anti-acetyl-lysine antibody at a dilution of 1:500 in the presence of 3% dry milk.
Immunoprecipitation with anti-CACT or anti-acetyl-lysine antibody
Five μg of anti-CACT or anti-acetyl-lysine antibody in PBS buffer were incubated for 2 h at 4 °C with protein G agarose beads for conjugation. Conjugated antibodies were then incubated with mitochondrial or mitoplast extract obtained by solubilization of mitochondria or mitoplasts (1 mg protein) with 1.5% Triton X-100 in PBS buffer and centrifugation at 16,000×g for 15 min (the supernatant was collected). After overnight incubation, beads were sedimented by centrifugation and washed with PBS buffer five times. Beads were then resuspended in 3× Laemmli buffer and boiled (5 min). Proteins were then used for analysis by western blotting and immunodecorated with anti-CACT or anti-acetyl-lysine antibodies, in parallel samples.
Overexpression and isolation of the recombinant rat CACT protein
Overexpression of rat CACT was obtained in E. coli and then the protein was purified, as previously described [15].
Reconstitution of native and recombinant rat CACT proteins in liposomes
Mitochondria were purified from rat liver by a protocol based on homogenization and centrifugation [20]. Mitoplasts were prepared as previously described [21]. Rat liver mitochondria were solubilized (0.3 mg proteins in 1.5% Triton X-100) and reconstituted into liposomes using a detergent removal method [22]. In this procedure, mixed micelles of phospholipids, detergent, and protein were passed several times through the same hydrophobic ion-exchange column. The reconstitution mixture proteins were composed of protein (about 150 µg solubilized mitochondrial protein), 1% Triton X-100, sonicated liposomes (10 mg phospholipids from egg yolk) [20], 10 mM Pipes pH 7.0, 15 mM carnitine, in 680 μl total volume. The prepared mixture was passed 15 times through a column (Pasteur pipette filled with 0.5 g XAD-4 Amberlite resin) pre-equilibrated with 10 mM Pipes pH 7.0, 15 mM carnitine. Passages through the column were performed at room temperature. The same method was also used for reconstituting the recombinant rat CACT (about 0.6 μg protein).
Transport measurements
Proteoliposomes obtained as described in “Reconstitution of native and recombinant rat CACT proteins in liposomes” section were used for transport assay. To this aim, aliquots (550 μl) of proteoliposomes were chromatographed on Sephadex G-75 column (15 cm height, 0.75 cm internal diameter) for removing substrate from the external compartment. Six hundred microliters containing the proteoliposomes (turbid eluate) were collected, and divided in aliquots of 100 μl for transport assay. Uptake assay was started by 0.1 mM [3H]carnitine addition. At the indicated time, 1.5 mM NEM (CACT inhibitor) was added to terminate the transport reaction. The inhibitor was added together with the [3H]carnitine (time zero) to perform control samples. After transport had been stopped, the external [3H]carnitine was removed by Sephadex G-75 chromatography, and the intraliposomal radioactivity was counted by a β-counter after addition of a scintillation cocktail. Transport values were obtained after subtracting control values (with inhibitor added at time zero).
Results
Identification of acetylated CACT in mitochondria
As previously described, the CACT can be immunoprecipitated from the mitochondrial extract by an in house produced polyclonal antibody [23]. As shown by Fig. 1, the immunoprecipitated CACT is detected by the same anti-CACT antibody as a band at about 32.5 kDa (Fig. 1a), corresponding to the molecular mass of CACT [15]. The protein is also stained by an anti-acetyl-lys antibody (Fig. 1b), indicating that at least a fraction of the transporter molecules is acetylated. The CACT can also be immunoprecipitated by the anti-acetyl-lys antibody, as demonstrated by staining of this preparation with anti-CACT antibody (Fig. 1c). These data confirm the presence of acetylated CACT in mitochondria. Some bands at higher molecular mass are immunostained together with the CACT monomer; these bands might correspond to the heavy chain of the antibody or to a dimeric form of CACT. To verify whether CACT could be enzymatically deacetylated, CACT, extracted from mitochondria as in the experiment of Fig. 1, was incubated with SIRT3. After immunoprecipitation, no apparent variation of staining with the anti-acetyl-lys antibody was observed (not shown). It is known that deacetylation mediated by SIRT3, in vivo, is dependent on NAD+ [24]. Therefore, the SIRT3 treatment was performed together with NAD+, in the presence or absence of the SIRT inhibitor NAM [24], and immunostained with anti-acetyl-lysine or with anti-CACT antibody, as control. Pretreatment of solubilized mitochondria with SIRT3, NAD+, and NAM before immunoprecipitation resulted in a band immunostained by the anti-acetyl-lys antibody, similar to the untreated control (Fig. 2a, lanes 1 and 2). Upon treatment of the CACT with SIRT3 in the presence of NAD+ but not of the inhibitor (Fig. 2a, lane 3), immunostaining with the anti-acetyl-lys was virtually abolished. In all the lanes, i.e., without or with treatments, the protein was clearly recognized by the anti-CACT antibody indicating the presence of comparable protein amounts (Fig. 2a, lower panel). This result indicated that the CACT was mostly deacetylated by SIRT3 in the presence of NAD+ and that the deacetylation reaction was inhibited by NAM. To ascertain whether the expected acetylation sites protrude towards the mitochondrial matrix or the intermembrane space of mitochondria, rat liver mitoplasts (mitochondria without outer membrane) were treated with SIRT3. After washing, mitoplasts were solubilized and the protein extract subjected to immunoprecipitation with anti-CACT antibody. Figure 2b shows the results obtained by immunostaining with both antibodies. The data demonstrates that CACT was still acetylated after treatment of intact mitoplasts with SIRT3, i.e., the enzyme could not reach the acetylation site(s) (Fig. 2b, lane 1). As a control, mitoplasts were solubilized before treatment with SIRT3. In this case, the protein was deacetylated (Fig. 2b, lane 2), as expected and in agreement with data in Fig. 2a. The presence of comparable amounts of CACT under both the conditions was tested by the anti-CACT antibody (Fig. 2b, lower panel). Therefore, the acetylated lys residues are located in a protein moiety, which is not accessible from the intermembrane space, i.e., these residues are exposed towards the matrix. The results are also in agreement with the acetylome of PhosphoSitePlus database [12], indicating acetylation of K148, K157, K170, and K244 of CACT on the basis of raw proteomic data. To gain further insights in the structure/function relationships of the acetylation of lys residues, the structural model of CACT has been employed. On the basis of the model, the protein is constituted by six hydrophobic α-helices crossing the membrane and surrounding a central hydrophilic cavity where the substrate binds to the residues R275, D179, R178, and H29. Hydrophilic α-helices h34 and h56 located in the matrix connect the hydrophobic α-helices H3–H4 and H5–H6 [13, 25, 26]. The lys residues K148, K157, and K244 are located in the hydrophilic loops h34 and h56 of the CACT which connects the α-helices H3–H4 and H5–H6, respectively; K170 is located at the matrix end of the α-helix H4 (Fig. 3a, b). These residues protrude towards the matrix and are far from the substrate binding site, whose residues are located at about the midpoint of the membrane, in the water-filled cavity. Moreover, the lys residues targets of acetylation are located on the matrix surface of the protein and protrudes towards the aqueous environment, as shown in Fig. 4a, b, thus being suitable for chemical modification by intramitochondrial acetyl-CoA. On the contrary, these residues are not accessible from the intermembrane space, as highlighted by the front view of the transporter showing the water-filled cavity where the residues responsible for substrate binding, but not responsible for acetylation, are visible (Fig. 4c).

Western blot analysis of acetylated CACT. Rat liver mitochondrial extract obtained as described in “Materials and methods” section was: a immunoprecipitated by anti-CACT and immunostained with the same antibody; b immunoprecipitated by anti-CACT and immunostained with anti-acetyl-lysine antibody; c immunoprecipitated by anti-acetyl-lysine and immunostained with anti-CACT antibody. The image is representative of at least two independent experiments. Migration of molecular mass standards is indicated by arrows

CACT deacetylation by SIRT3 and localization of acetylated sites. a Protein extract of rat liver mitochondria was incubated at 4 °C, 1 h with no enzyme (lane 1), 3 μg SIRT3 + 5 mM NAD+ + 10 mM NAM (lane 2), or 3 μg SIRT3 + 5 mM NAD+ (lane 3) and then immunoprecipitated with anti-CACT as described in “Materials and methods” section. Western blot analysis of the samples was performed with anti-acetyl-lysine (upper panel) or with anti-CACT (lower panel) antibodies, in parallel samples. b Intact mitoplasts (lane 1) or mitoplasts solubilized with Triton X-100 (lane 2) were incubated with 3 μg SIRT3 + 5 mM NAD+. Western blot analysis of mitoplast extract immunoprecipitated with anti-CACT and immunostained with anti-acetyl-lysine antibody (upper panel) or with anti-CACT antibody (lower panel), in parallel samples. The image is representative of at least two independent experiments. Migration of molecular mass standards is indicated by arrows

Structural model of acetylated residues of CACT. a Ribbon diagram of the CACT structural model from lateral or b from bottom (matrix) view. The homology model was constructed using the structure of the bovine ADP/ATP carrier [17] as template and the computer application Swiss PDB viewer as previously described [29]. The protein is constituted by six transmembrane α-helices depicted in gray-white, which surround a central cavity where the substrate binds. Matrix α-helix loops h34 and h56 (in dark gray) connect, respectively, α-helices H3–H4 and H5–H6. Mitochondrial inner membrane is represented by black lines. Highlighted residues are numbered. The residues of the active site are H29, R178, D179, and R275; the putative acetylation sites are K148, K157, K170, and K244

Surface exposure of acetylated residues of CACT. a Space-filled diagram of the CACT structural model from side (membrane) view, corresponding to the view of Fig. 3a. Mitochondrial inner membrane is represented by black lines. b Space-filled diagram of the CACT structural model from bottom (matrix side) view as highlighted by the cartoon. The residues of the putative acetylation sites are highlighted and numbered. c Space-filled diagram of CACT from top (cytosolic/intermembrane space side) view as highlighted by the cartoon; the residues of the active site are highlighted and numbered. The homology model was constructed as described in Fig. 3
Effect of acetylation on CACT function
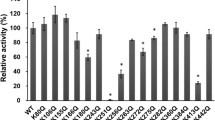
The possible consequences of acetylation on the CACT function have been investigated in proteoliposomes. Figure 5a shows the transport activity associated to the mitochondrial protein extract, pretreated or not with SIRT3 and then reconstituted in proteoliposomes. Transport was specifically measured as carnitine antiport (see “Materials and methods” section and Ref. [16]). Activity of samples pretreated with SIRT3 increased with respect to the control (Fig. 5a), indicating that acetylation of CACT impairs its function. The acetylated transporter exhibits a transport activity about half that of the deacetylated protein or of the protein treated with SIRT3 in the presence of NAD. Transport at equilibrium, which is proportional to the fraction of active proteoliposomes, i.e., liposomes harboring active transporters, is strongly impaired by acetylation. The resulting transport rate, calculated as the product of k (first order rate constant) and transport at equilibrium, is also impaired in the case of acetylated protein. The values are: 0.43 ± 0.094 or 1.04 ± 0.18 μmol min−1 g protein−1 for the acetylated or deacetylated protein, respectively. These data indicate that a fraction of the protein might be inactive. Kinetic analysis was performed by following transport rate of acetylated or deacetylated proteins in proteoliposomes, as dependence of substrate concentration. Data reported according to double reciprocal, plots (Fig. 5b), indicated that half-saturation constant of the protein for substrate was not substantially influenced by the PTM; their values were 0.36 ± 0.031 or 0.46 ± 0.10 mM, respectively. While the V max of the acetylated protein was about one-third (1.7 ± 0.20 µmol min−1 g protein−1) compared to that of deacetylated one (5.1 ± 0.75 µmol min−1 g protein−1). These data are again in agreement with that of Fig. 5a, indicating that the acetylated fraction of the protein is inactive. The behavior of acetylation is, indeed, analogous to that of a non-competitive inhibitor. As previously reported, acetylation could occur by non-enzymatic reaction of proteins with acetyl-CoA [7, 8]. To test possible occurrence of non-enzymatic acetylation, the recombinant CACT, which is acetyl-free, was employed (Fig. 6). Moreover, the recombinant CACT protein preparation does not contain any other contaminant that might influence the acetylation reaction. After incubation of CACT with acetyl-CoA and reconstitution in proteoliposomes, transport activity decreased from about 25% at equilibrium (Fig. 6a). Transport rates were calculated as the product of k (first order rate constant) and transport at equilibrium in the two different conditions. Transport rate of the acetylated protein was also slightly influenced being 250 ± 32 or 280 ± 43 µmol min−1 g protein−1, respectively, for acetylated or not acetylated protein. By dose–response analysis, the IC 50 of the transporter for acetyl-CoA resulted to be 5.5 mM (not shown). It has to be stressed that, as expected, the specific activity of the purified recombinant protein is much higher than that extracted from mitochondria [10]. Occurrence of acetylation was then checked by western blot analysis with the anti-acetyl-lys antibody. As shown in Fig. 6b, an immunostained band appeared at the molecular mass of CACT, after incubation with acetyl-CoA. While no reaction was observed in the control lane corresponding to the CACT not incubated with acetyl-CoA, the presence of CACT protein in both lanes is shown, in the lower panel, by immunodetection of corresponding samples with the anti-CACT antibody.

Effect of CACT acetylation on transport function. a Transport was measured as described in “Materials and methods” section adding measuring [3H]carnitine/carnitine antiport in proteoliposomes reconstituted with mitochondrial extract incubated in the absence (white circle) or in the presence of SIRT3 + NAD+ + NAM (black circle) or SIRT3 + NAD+ (white square). In black circle and white square the reagents were added to the solubilized mitochondria for 1 h at 4 °C before the re constitution procedure. The transport reaction was stopped at the indicated times, as described in “Materials and methods” section. Reported values are mean ± SD from three experiments. b Kinetic analysis of the effect of SIRT3 on the reconstituted CACT. The [3H]carnitine/carnitine antiport rate was measured, as described in “Materials and methods” section, adding [3H]carnitine at different concentrations to proteoliposomes reconstituted with mitochondrial extract and containing 15 mM carnitine, in the absence (white circle) or in the presence of (dark circle) SIRT3 + NAD+. In dark circle the reagents were added to the solubilized mitochondria for 1 h at 4 °C before the reconstitution procedure. Experimental data plotted according to Lineweaver–Burk as reciprocal transport rate versus reciprocal carnitine concentrations. Reported values are mean ± SD from three experiments

Non-enzymatic acetylation of recombinant rat CACT. a Time course of carnitine transport activity assayed as described in “Materials and methods” section after incubation of recombinant CACT with (dark circle) or without (white circle) 2.5 mM acetyl-CoA for 3 h at 37 °C under shaking. Reported values are mean ± SD from three experiments. b Upper panel immunodecoration by anti-acetyl-lysine antibody of: untreated CACT (lane 1), CACT pretreated with acetyl-CoA (lane 2). Lower panel immunodecoration of the same samples after blotting on a separate membrane, with anti-CACT antibody. The image is representative of at least two independent experiments. Migration of molecular mass standards is indicated by arrows
Discussion
The described results highlight that a fraction of CACT extracted from rat liver mitochondria is acetylated. Acetylation, indeed, is a recently recognized wide phenomenon in mitochondria which is involved in regulation of several pathways. Some components of the β-oxidation pathway resulted acetylated in previous studies focused to the analysis of the acetylome [1]. Among the most well described, there is the LCAD whose acetylation is involved in the control of the enzyme function. LCAD activity is decreased by acetylation thus decreasing the β-oxidation flux. This PTM has been recognized as a novel regulatory mechanism for mitochondrial β-oxidation [1, 9]. Acetylation of CACT causes decrease of transport activity correlating well with the control of LCAD by the same PTM. The CACT was previously described to be regulated by physiological factors such as the GSH/GSSG balance [10], the H2S level [11], or by the production of ROS (H2O2), causing CACT sulfenylation [27]. These factors influence the redox state of CACT via two specific Cys residues, C136 and C155. Thus, CACT is considered as a possible regulatory site of β-oxidation pathway. Acetylation plays an additional role in the control of CACT function which, together with the LCAD, contributes to the regulation of β-oxidation. This mechanism represents a control of the influx of fatty acyl units into mitochondria in response to intramitochondrial acetyl-CoA level, in addition to the regulation of CPT1 by malonyl-CoA. The more likely acetylated residues of CACT are indicated by proteomic analyses [12] (Fig. 3). These residues which protrude towards the aqueous environment of the mitochondrial matrix should be available to acetylation by both enzymatic and non-enzymatic mechanisms. The experiments performed with the protein extracted from mitochondria validate all the data obtained by modeling. Moreover, the functional data allow us to hypothesize that the activity impairment caused by acetylation is due to a steric hindrance exerted by the acetyl groups bound to the lys residues (Fig. 3a). These groups may impair conformational changes of the transporter rather than binding of substrate, as demonstrated by the kinetic analysis of transport of acetylated and deacetylated proteins. Interestingly, the effect (inhibition) of acetylation on CACT is opposite with respect to that described for the citrate carrier (CIC) which is activated by the same PTM [6]. This difference is, however, in agreement with the involvement of the two transporters in opposite metabolic pathways, i.e., the CACT is involved in β-oxidation, while the CIC is involved in biosynthesis of fatty acids. Thus, under conditions of increased acetyl-CoA level and protein acetylation in mitochondria, fatty acid synthesis is stimulated while β-oxidation is depressed by PTM of the two transporters. This regulatory mechanism will prevent further acetyl-CoA formation facilitating the release of free CoA in mitochondria. The mechanism of acetylation of CACT is in line with a dynamic control of the protein which could undergo an on/off switch caused by deacetylation/acetylation linked to the availability of acetyl-CoA. Indeed, the acetyl-CoA pool is also linked to the acetyl-carnitine availability which represents a buffer for intramitochondrial acetyl-CoA [7]. Acetyl-carnitine level is in turn regulated by the activity of the CACT, since acetyl-carnitine is transported in exchange for free carnitine [16]. Thus far, while acetylation of mitochondrial proteins seems a general mechanism of control, in the case of the CACT it may result as a specific mechanism with a feedback control represented by the acetyl-CoA/acetyl-carnitine interconversion mediated by the mitochondrial Carnitine O-acetyltransferase (CAT) [7, 28]. The CACT results an additional mitochondrial component which can be non-enzymatically acetylated under conditions of excess mitochondrial acetyl-CoA, by mass action, as recently highlighted for many mitochondrial proteins [7], given that the concentration of acetyl-CoA is relatively high in mitochondria (about 5 mM [30]). In this scenario, the CACT, together with other proteins, may be a target of pathological acetylation caused by impaired acetyl-CoA buffering.
Abbreviations
- SIRT:
-
Sirtuin
- BSA:
-
Bovine serum albumin
- PTM:
-
Post-translational modification
- CACT:
-
Carnitine/acylcarnitine transporter
- LCAD:
-
Long-chain acyl-CoA dehydrogenase
- CPT1:
-
Carnitine palmitoyltransferase I
- CAT:
-
Carnitine O-acetyltransferase
- GSH:
-
l-Glutathione reduced
- GSSG:
-
l-Glutathione oxidized
- AAC:
-
ADP/ATP carrier
- NAM:
-
Nicotinamide
- PVDF:
-
Polyvinyldifluoride membrane
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NEM:
-
N-Ethylmaleimide
References
Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327:1000–1004. doi:10.1126/science.1179689
Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327:1004–1007. doi:10.1126/science.1179687
Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23:607–618. doi:10.1016/j.molcel.2006.06.026
Rauh D, Fischer F, Gertz M, Lakshminarasimhan M, Bergbrede T, Aladini F, Kambach C, Becker CF, Zerweck J, Schutkowski M, Steegborn C (2013) An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat Commun 4:2327. doi:10.1038/ncomms3327
Weinert BT, Schölz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C (2013) Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep 4:842–851. doi:10.1016/j.celrep.2013.07.024
Palmieri EM, Spera I, Menga A, Infantino V, Porcelli V, Iacobazzi V, Pierri CL, Hooper DC, Palmieri F, Castegna A (2015) Acetylation of human mitochondrial citrate carrier modulates mitochondrial citrate/malate exchange activity to sustain NADPH production during macrophage activation. Biochim Biophys Acta 1847:729–738. doi:10.1016/j.bbabio.2015.04.009
Davies MN, Kjalarsdottir L, Thompson JW, Dubois LG, Stevens RD, Ilkayeva OR, Brosnan MJ, Rolph TP, Grimsrud PA, Muoio DM (2016) The acetyl group buffering action of carnitine acetyltransferase offsets macronutrient-induced lysine acetylation of mitochondrial proteins. Cell Rep 14:243–254. doi:10.1016/j.celrep.2015.12.030
Pougovkina O, te Brinke H, Ofman R, van Cruchten AG, Kulik W, Wanders RJ, Houten SM, de Boer VC (2014) Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum Mol Genet 23:3513–3522. doi:10.1093/hmg/ddu059
Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Alt FW, Kahn CR, Verdin E (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–125. doi:10.1038/nature08778
Giangregorio N, Palmieri F, Indiveri C (2013) Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim Biophys Acta 1830:5299–5304. doi:10.1016/j.bbagen.2013.08.003
Giangregorio N, Tonazzi A, Console L, Lorusso I, De Palma A, Indiveri C (2016) The mitochondrial carnitine/acylcarnitine carrier is regulated by hydrogen sulfide via interaction with C136 and C155. Biochim Biophys Acta 1860:20–27. doi:10.1016/j.bbagen.2015.10.005
Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43:D512–D520. doi:10.1093/nar/gku1267
Tonazzi A, Giangregorio N, Console L, Indiveri C (2015) Mitochondrial carnitine/acylcarnitine translocase: insights in structure/function relationships. Basis for drug therapy and side effects prediction. Min Rev Med Chem 15:396–405
Giangregorio N, Tonazzi A, Indiveri C, Palmieri F (2007) Conformation-dependent accessibility of Cys-136 and Cys-155 of the mitochondrial rat carnitine/acylcarnitine carrier to membrane-impermeable SH reagents. Biochim Biophys Acta 1767:1331–1339. doi:10.1016/j.bbabio.2007.08.010
Indiveri C, Iacobazzi V, Giangregorio N, Palmieri F (1998) Bacterial overexpression, purification, and reconstitution of the carnitine/acylcarnitine carrier from rat liver mitochondria. Biochem Biophys Res Commun 249:589–594. doi:10.1006/bbrc.1998.9197
Giangregorio N, Console L, Tonazzi A, Palmieri F, Indiveri C (2014) Identification of amino acid residues underlying the antiport mechanism of the mitochondrial carnitine/acylcarnitine carrier by site-directed mutagenesis and chemical labeling. Biochemistry 53:6924–6933. doi:10.1021/bi5009112
Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ, Brandolin G (2003) Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426:39–44. doi:10.1038/nature02056
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Tonazzi A, Mantovani C, Colella M, Terenghi G, Indiveri C (2013) Localization of mitochondrial carnitine/acylcarnitine translocase in sensory neurons from rat dorsal root Ganglia. Neuro Chem Res 38:2535–2541. doi:10.1007/s11064-013-1168-z
Indiveri C, Capobianco L, Krämer R, Palmieri F (1989) Kinetics of the reconstituted dicarboxylate carrier from rat liver mitochondria. Biochim Biophys Acta 977:187–193
Kaplan RS, Pedersen PL (1985) Isolation and reconstitution of the n-butylmalonate-sensitive dicarboxylate transporter from rat liver mitochondria. J Biol Chem 260:10293–10298
Scalise M, Pochini L, Giangregorio N, Tonazzi A, Indiveri C (2013) Proteoliposomes as tool for assaying membrane transporter functions and interactions with xenobiotics. Pharmaceutics 5:472–497. doi:10.3390/pharmaceutics5030472
Console L, Giangregorio N, Indiveri C, Tonazzi A (2014) Carnitine/acylcarnitine translocase and carnitine palmitoyltransferase 2 form a complex in the inner mitochondrial membrane. Mol Cell Biochem 394:307–314. doi:10.1007/s11010-014-2098-z
Verdin E, Hirschey MD, Finley LW, Haigis MC (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35:669–675. doi:10.1016/j.tibs.2010.07.003
Giangregorio N, Tonazzi A, Console L, Indiveri C, Palmieri F (2010) Site-directed mutagenesis of charged amino acids of the human mitochondrial carnitine/acylcarnitine carrier: insight into the molecular mechanism of transport. Biochim Biophys Acta 1797:839–845. doi:10.1016/j.bbabio.2010.03.017
Tonazzi A, Giangregorio N, Indiveri C, Palmieri F (2009) Site-directed mutagenesis of the His residues of the rat mitochondrial carnitine/acylcarnitine carrier: implications for the role of His-29 in the transport pathway. Biochim Biophys Acta 1787:1009–1015. doi:10.1016/j.bbabio.2009.02.026
Tonazzi A, Console L, Indiveri C (2013) Inhibition of mitochondrial carnitine/acylcarnitine transporter by H(2)O(2): molecular mechanism and possible implication in pathophysiology. Chem Biol Interact. doi:10.1016/j.cbi.2013.01.006
Arduini A, Bonomini M, Savica V, Amato A, Zammit V (2008) Carnitine in metabolic disease: potential for pharmacological intervention. Pharmacol Ther 120:149–156. doi:10.1016/j.pharmthera.2008.08.008
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. doi:10.1002/elps.1150181505
Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, Gibson BW (2013) Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci 110(16):6601–6606. doi:10.1073/pnas.1302961110
Acknowledgements
This work was supported by funds from: Programma Operativo Nazionale [01_00937] - MIUR “Modelli sperimentali biotecnologici integrati per lo sviluppo e la selezione di molecole di interesse per la salute dell’uomo”.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The experiments comply with the current Italian laws.
Additional information
Nicola Giangregorio and Annamaria Tonazzi have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Giangregorio, N., Tonazzi, A., Console, L. et al. Post-translational modification by acetylation regulates the mitochondrial carnitine/acylcarnitine transport protein. Mol Cell Biochem 426, 65–73 (2017). https://doi.org/10.1007/s11010-016-2881-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2881-0