
Abstract
The cytoprotective role of heat shock proteins (HSPs) has been demonstrated in various cell types however, only few studies have investigated the role of extracellular exposure to HSPs in the survival of human lymphoma cell line U937. In the present study, we investigated the effect of extracellular exposure to four HSPs (HSP90, HSP70, HSP60, and HSP47) on apoptotic cell death induced by either oxidative stress (hydrogen peroxide) or endoplasmic reticulum stress-mediated intracellular calcium overload. It was found that extracellular exposure to HSPs reduced the cytotoxicity induced by hydrogen peroxide, but not that evoked by thapsigargin (a specific inhibitor of cytosolic calcium reuptake which is able to induce endoplasmic reticulum stress with subsequent intracellular calcium overload). Similarly, it was observed that exogenous HSPs were able to suppress the caspase-3 activation induced by hydrogen peroxide. These findings indicate that extracellular HSPs increase the resistance of human lymphoma cell line U937 to apoptotic cell death induced by hydrogen peroxide and diminish oxidative stress-mediated injures.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heat shock proteins (HSPs) are an evolutionarily conserved superfamily of proteins [1]. As their name suggests, HSPs were originally discovered to be upregulated after exposure to cells to elevated temperatures; however, they are also induced in response to a variety of other stress stimuli [2]. Mammalian HSPs have been classified into two groups according to their size: high molecular weight HSPs and small molecular weight HSPs. The first group includes three major families: HSP90 (HSPC), HSP70 (HSPA), and HSP60 (HSPD), which are ATP-dependent chaperones. By contrast, small HSPs (sHSPs or HSPB), such as HSP27, are ATP-independent chaperones [3].
Apoptosis is a gene-regulated form of cell death that is critical for normal development and tissue homeostasis. A major component of the apoptotic machinery involves a family of aspartic acid-directed cysteine proteases, called caspases (cysteinyl aspartate-specific proteinases), which cleave multiple protein substrates en masse, thereby leading to the loss of cellular structure and function and ultimately resulting in cell death [4].
Many different cell death stimuli are well-known inducers of HSPs expression. Additionally, accumulating data clearly indicate that HSPs have essential anti-apoptotic properties. Thus, HSPs can block both the intrinsic and the extrinsic apoptotic pathways through the interaction with essential proteins at three levels: (i) upstream the mitochondria level, thereby modulating signaling pathways; (ii) at the mitochondrial level, controlling the release of apoptogenic molecules; (iii) and the post-mitochondrial level, by blocking apoptosis from a later phase than any known survival-enhancing drug or protein [5–7]. When associated with key stress signaling pathways and apoptotic molecules, HSPs block cell death and promote survival and proliferation [8]. In fact, HSPs have been shown to block apoptosis by interfering with caspase activation. Besides, overexpression of HSP27, HSP70, HSP60, or HSP90, inhibits apoptosis and prevents caspase activation in many different cellular models upon a variety of cellular stresses [9–11]. On the contrary, depletion of HSP27, HSP60, HSP70, or HSP90, either by anti-sense constructions or siRNA strategies, increases the cell sensitivity to apoptotic stimuli [12, 13].
Growing evidence suggests that HSPs can be released into the extracellular space and can have a wide variety of extracellular functions. For instance, release of HSP72 has been demonstrated in human peripheral blood mononuclear cells, human macrophages, human epithelial cells, and human tumor cells [14]. In the nervous system, HSP72 is released from astrocytes or Schwann cells and can affect neighboring neuron/axon [15].
Despite the fact that the cytoprotective role of HSPs has been demonstrated in various cell types, it is little known whether extracellular HSPs play the same protective role in lymphoma cells. This study was designed to investigate whether exogenous HSPs have protective effects against two models of apoptotic cell death induced by intracellular calcium overload or oxidative stress (hydrogen peroxide, H2O2) in human lymphoma cell line U937.
Materials and methods
Chemicals and reagents
U937 cell line (ECACC No 85011440) derived from human histiocytic lymphoma was purchased from The European Collection of Cell Cultures (ECACC) (Dorset, UK). Fetal bovine serum (FBS) and penicillin/streptomycin were obtained from HyClone (Aalst, Belgium). l-Glutamine and RPMI 1640 medium were purchased from Cambrex (Verviers, Belgium). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Sigma (Madrid, Spain). HSP90, HSP70, HSP60, and HSP47 were acquired from Enzo Life Sciences (Madrid, Spain). Anti-HSP90 (F-8) and anti-HSP70 (N27F3-4) were purchased from Santa Cruz Biotechnology, Inc. (Heidelberg, Germany). Anti-β-actin (8H10D10) antibody was procured from Cell Signaling (Danvers, MA, USA). All other reagents were of analytical grade.
Cell culture
U937 cells were derived from malignant cells of a pleural effusion of a 37-year-old Caucasian male with diffuse histiocytic lymphoma. U937 cells (passages 6–12) were grown in RPMI 1640 medium supplemented with 2 mM l-glutamine, 10 % heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37 °C under a humidified condition of 95 % air and 5 % CO2. Cells were routinely plated into 24-well plates at a density of 3 × 105 cells/mL, and the viability was >95 % in all experiments as assayed by the trypan-blue exclusion method.
Study groups
U937 cells were treated with different concentrations (100, 10, and 1 ng/mL) of HSP90, HSP70, HSP60, and HSP47, or left unstimulated (control), for 3 h. Subsequently, cells were challenged with 2.5 µM thapsigargin or 60 µM H2O2 for 24 h. The dosage and incubation times of HSPs, thapsigargin, and H2O2 have been previously used in other studies (1, 10, 16, 18, 19).
Cell viability assay
Cell viability was evaluated using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, which is based on the ability of viable cells to convert a water-soluble, yellow tetrazolium salt into a water-insoluble, purple formazan product. The enzymatic reduction of the tetrazolium salt happens only in living, metabolically active cells, but not in dead cells. Cells were seeded in 24-well plates at a density of 2 × 105 cells per well, and subsequently, exposed to the appropriate treatment at 37 °C. After the treatments, the medium was removed, and MTT was added into each well, and then incubated for 60 min at 37 °C, as previously described [16]. The supernatant was discarded, and DMSO was added to dissolve the formazan crystals. Treatments were carried out in triplicate. Optical density was measured in an automatic microplate reader (Infinite M200, Tecan Austria GmbH, Groedig, Austria) at a test wavelength of 490 nm and a reference wavelength of 650 nm to nullify the effect of cell debris. Data are presented as percentage above control (untreated samples).
Assay for caspase activity
To determine caspase-3 activity, stimulated or resting cells were sonicated, and the cell lysates were incubated with 2 mL of substrate solution (20 mM HEPES, pH 7.4, 2 mM EDTA, 0.1 % CHAPS, 5 mM DTT, and 8.25 µM of caspase substrate) for 1 h at 37 °C as previously described [17]. The activity of caspase-3 was calculated from the cleavage of the specific fluorogenic substrate (AC-DEVD-AMC). Substrate cleavage was measured with an automatic microplate reader (Infinite M200) with excitation wavelength of 360 nm and emission at 460 nm. Preliminary experiments reported that caspase-3 substrate cleaving was not detected in the presence of the inhibitor of caspase-3, DEVD-CMK. Data were calculated as fluorescence units/mg protein and presented as fold-increase over the pre-treatment level (experimental/control).
Western blotting
One-dimensional sodium dodecyl sulfate (SDS) electrophoresis was performed with a 10 % Tris–glycine gel, and the separated proteins were then electrophoretically transferred onto nitrocellulose membranes for subsequent probing, in a semi-dry blotter for 2 h at 0.8 mA/cm2. Blots were incubated overnight with 5 % (w/v) nonfat dry milk in Tris-buffered saline with 0.1 % Tween 20 (Tris-buffered saline with Tween 20 (TBST)) to block residual protein-binding sites. Blocked membranes were then incubated for 3 h with anti-HSP90 (F-8), anti-HSP70 (N27F3-4), or anti-β-actin (8H10D10) antibodies diluted 1:500, 1:250, and 1:1000, respectively, in TBST. The primary antibody was removed, and blots were washed three times for 10 min each with TBST. To detect the primary antibodies, blots were incubated with the appropriate horseradish peroxidase-conjugated anti-IgG antibody diluted 1:5000 in TBST, washed three times in TBST, and exposed to enhanced chemiluminescence reagents for 5 min. Blots were then exposed to photographic films, and the optical density was estimated using scanning densitometry.
Statistical analysis
Data are presented as mean ± standard error of mean (S.E.M) for each group. To compare the different treatments, statistical significance was calculated by one-way analysis of variance (ANOVA) followed by post hoc Tukey test. P < 0.05 was considered to indicate a statistically significant difference.
Results
First, it was determined whether HSP90 and HSP70 protein levels are modified upon treatment with the reactive oxygen species H2O2 or thapsigargin, a specific inhibitor of cytosolic calcium reuptake which is able to induce endoplasmic reticulum stress, in U937 cell line. In previous studies, our results have demonstrated that both H2O2 and thapsigargin induce caspase-dependent apoptosis and cytotoxicity effects [18, 19]. As shown in Fig. 1, Western blot analysis revealed that treatment of U937 cells with 60 µM H2O2 or 2.5 µM thapsigargin for 24 h significantly decreased the expression levels of both HSP90 and HSP70 compared to control values (untreated cells) (Fig. 1).

Both thapsigargin and hydrogen peroxide modify HSP intracellular content. To determine HSP90 and HSP70 content, U937 cells were stimulated with thapsigargin (TGS; 2.5 µM) or hydrogen peroxide (H2O2; 60 µM) for 24 h. Samples were lysed and then subjected to gradient Tris–glycine isolation and subsequent Western blotting with a specific anti-HSP90 (F-8) or anti-HSP70 (N27F3-4) antibody, and reprobed with anti-β-actin (8H10D10) antibody for protein loading control. a Blots representative of five independent experiments. b Densitometric analysis showing the relative content of HSP90 and HSP70. *P < 0.05 compared to control values
In order to examine the effect of extracellular HSPs on cell viability, U937 cells were incubated with different concentrations (100, 10, and 1 ng/mL) of HSP90, HSP70, HSP60, and HSP47 for 3 h. As shown in Fig. 2, HSPs induced a small, nonsignificant decrease on cell viability of U937 cell line. Only HSP60, at 100 and 10 ng/mL, produces statistically significant (P < 0.05) decrease in cell viability (Fig. 2c). Parallel assays were performed to determine whether administration of HSPs can provide protective effects in U937 cells when exposed to H2O2 or thapsigargin. As expected, incubation of cells for 24 h with 2.5 μM thapsigargin (Fig. 3) and 60 μM H2O2 (Fig. 4) induced a significant decrease in cell viability (42.28 ± 3.75 and 65.85 ± 4.75 % of control, P < 0.05, respectively). However, when U937 cells were pre-treated with different concentrations (100, 10, and 1 ng/mL) of HSP90, HSP70, HSP60, or HSP47 for 3 h and then exposed to 60 µM H2O2, cell viability increased compared to H2O2 treatment alone, although it was not clearly concentration dependent (Fig. 4). This effect was statistically significant (P < 0.05) when cells were pre-treated with HSPs at 1 to 10 ng/mL. These results indicated that the administration of HSPs exerted considerable protective effects against H2O2-induced cytotoxicity in U937 cell line. However, HSPs, at any of the doses tested, were unable to reverse the decrease in cell survival induced by thapsigargin (Fig. 3).

Dose–response effect of different HSPs on U937 cell viability. Cells were incubated with different concentrations (100, 10, and 1 ng/mL) of HSP90 (a), HSP70 (b), HSP60 (c), and HSP47 (d), or unstimulated (control), for 3 h. Cell viability was estimated as described under Materials and methods. Values are presented as mean ± SEM of six separate experiments and expressed as percentage of control values (untreated samples). *P < 0.05 compared to control values

HSPs showed no effect on thapsigargin-induced reduction of U937 cell viability. Cells were pre-incubated with decreasing concentrations (100, 10, and 1 ng/mL) of HSP90 (a), HSP70 (b), HSP60 (c), and HSP47 (d) or the vehicle for 3 h, and then stimulated with thapsigargin (TGS; 2.5 µM) for 24 h. Cell viability was estimated as described under Materials and methods. Values are presented as mean ± SEM of six separate experiments and expressed as percentage of control values (untreated samples). *P < 0.05 compared to control values

Low concentrations of HSPs reversed the effect of hydrogen peroxide on U937 cell viability. Cells were pre-incubated with decreasing concentrations (100, 10, and 1 ng/mL) of HSP90 (a), HSP70 (b), HSP60 (c), and HSP47 (d) or the vehicle for 3 h, and then stimulated with hydrogen peroxide (H2O2; 60 µM) for 24 h. Cell viability was estimated as described under Materials and methods. Values are presented as mean ± SEM of six separate experiments and expressed as percentage of control values (untreated samples). *P < 0.05 compared to control values. # P < 0.05 compared to cell treated with H2O2 alone
In subsequent experiments, we assessed whether the decrease observed in cell viability could be due to apoptotic cell death. For this purpose, the enzymatic activity of caspase-3 was determined. This protease is considered to be one of the major mediators of apoptotic cell death and its activation is by far considered an apoptotic marker. As shown in Figs. 5 and 6, treatment of cells with 1 ng/mL (the most effective dose in reversing H2O2-induced cytotoxicity) HSP90, HSP70, HSP60, and HSP47 for 3 h exerted negligible effects on caspase-3 activity. Additionally, treatment of U937 cells for 24 h with 2.5 µM thapsigargin (Fig. 5) or 60 µM H2O2 (Fig. 6) caused a significant activation of caspase-3 (2.68 ± 0.19 and 1.64 ± 0.03 fold-increase, P < 0.05, respectively). Yet again, when U937 cells were pre-incubated with 1 ng/mL HSP90, HSP70, HSP60, or HSP47 for 3 h, the increase in caspase-3 activity induced by 60 µM H2O2 was clear and significantly (P < 0.05) reverted compared to the results obtained with H2O2 alone (Fig. 6). Likewise, none of the doses of HSP tested were able to inhibit the stimulatory effect of 2.5 µM thapsigargin on the enzymatic activity of caspase-3 (Fig. 5).

HSPs displayed mild effect on thapsigargin-evoked caspase-3 activation in U937 cells. Cells were pre-incubated with 1 ng/mL HSP90 (a), HSP70 (b), HSP60 (c), and HSP47 (d) or the vehicle for 3 h, and then stimulated with thapsigargin (TGS; 2.5 µM) for 24 h. Caspase-3 activity was estimated as described under Materials and methods. Values are presented as mean ± SEM of seven separate experiments and expressed as fold-increase over the pre-treatment level (experimental/control). *P < 0.05 compared to control values

Low concentrations of HSPs counteracted hydrogen peroxide-stimulated caspase-3 activation in U937 cells. Cells were pre-incubated with 1 ng/mL HSP90 (a), HSP70 (b), HSP60 (c), and HSP47 (d) or the vehicle for 3 h, and then stimulated with hydrogen peroxide (H2O2; 60 µM) for 24 h. Caspase-3 activity was estimated as described under Materials and methods. Values are presented as mean ± SEM of seven separate experiments and expressed as fold-increase over the pre-treatment level (experimental/control). *P < 0.05 compared to control values. # P < 0.05 compared to H2O2-treated cells in the presence of the corresponding HSP
Discussion
The results of the present study suggest that extracellular exposure to four different HSPs, such as HSP90, HSP70, HSP60, and HSP47, reduced apoptotic cell death induced by the reactive oxygen species H2O2 in human lymphoma cell line U937. However, any of the four HSPs did not affect the cytotoxic effect of thapsigargin, a specific inhibitor of cytosolic calcium reuptake which is able to induce endoplasmic reticulum stress and intracellular calcium overload.
In previous studies, our findings have shown that both H2O2 and thapsigargin are able to induce apoptotic cell death, including caspase activation and cytotoxicity [18, 19]. Additionally, it has been shown that HSPs have cytoprotective and anti-apoptotic effects [for review see 20, 21]. Herein, it has been also demonstrated that the treatment of U937 cells with H2O2 or thapsigargin for 24 h significantly decreased the expression levels of both HSP90 and HSP70. Therefore, it is reasonable to point out that the pro-apoptotic effects of H2O2 and thapsigargin may be mediated, at least in part, by its ability to decrease expression levels of HSPs.
Early works with HSPs were primarily focused on their intracellular effects. More recently, interest has grown on the role of HSPs as extracellular signals contributing to the host response to injury or infection. Recombinant HSP72 has been shown to induce inflammatory cytokine release including tumor necrosis factor-α, interleukin-1, and interleukin-6 in monocytes and macrophages [21–23]. Moreover, the protective activity of extracellular HSP72 has been demonstrated in vivo and in vitro in the neuronal model system [14, 24, 25].
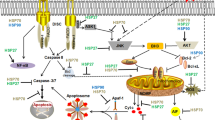
Oxidative stress, which is characterized by excessive ROS production, is one of several mechanisms that induce apoptosis in cells and has been proposed to be the cause of degenerative diseases [26–28]. Among a great variety of ROS, H2O2 plays a pivotal role, because it is generated from nearly all sources of oxidative stress and diffuses freely in and out the cells and tissues [29]. In the present study, our results indicated that H2O2 caused apoptotic cell death in U937 cell line, which was clearly inhibited by HSPs pre-treatment. General mechanism of apoptotic cell death induced by H2O2 and its relationship with HSPs are schematically described in Fig. 7. These results agree with previous studies reporting that extracellular HSP72 protects primary cultures of Schwann cells from H2O2-induced apoptosis [14], as well as with those claiming that heat shock transcription factor 1 was able to inhibit H2O2-induced cardiomyocyte death [30].

Schematic diagram describing the effects of H2O2 on apoptotic cells death
Caspases are a family of cysteine aspartic acid protease that are central regulators of apoptosis and form a proteolytic cascade, resulting in the cleavage of distinct and vital proteins. Caspase-3, a biochemical hallmark of apoptosis, is a critical effector caspase within apoptotic process. Our results demonstrate that the four HSPs tested prevent caspase-3 activation induced by H2O2 in U937 cells, resulting in suppression of H2O2-induced apoptosis. It is worth noticing that the inhibitory effect of HSPs on the decrease in cell viability was greater with the dose of 1 ng/mL compared with higher doses. We do not have a convincing explanation for this contradictory fact but it could be somehow attributable to desensitizing aspects of U937 cells when exposed to high concentrations of HSPs. Finally, our data also indicate that HSPs were unable to reverse the endoplasmic reticulum stress-induced apoptotic cell death evoked by thapsigargin, with subsequent intracellular calcium overload. These results agree with others obtained previously in human epidermoid carcinoma A-431 cells, which showed that alterations in intracellular calcium homeostasis resulted in diminished HSP72 mRNA production and less HSP72 synthesis [31].
In conclusion, the findings of this study have demonstrated that extracellular exposure to four different HSPs, such as HSP90, HSP70, HSP60, and HSP47, inhibited H2O2-induced apoptotic cell death without affecting apoptosis process evoked by intracellular calcium overload. The results presented herein support the hypothesis that exogenous HSPs are physiologically similar to endogenous HSPs, thereby functioning in a similar manner in oxidatively stressed cells and having similar protective activities.
References
Garrido C, Gurbuxani S, Ravagnan L, Kroemer G (2001) Heat shock proteins: endogenous modulators of apoptotic cell death. Biochem Biophys Res Commun 286:433–442
Morimoto RI (1998) Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 12:3788–3796
Parceiller A, Gurbuxani S, Schmitt E, Solary E, Garrido C (2003) Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem Biophys Res Commun 304:505–512
Stennicke HT, Salvesen GS (1997) Biochemical characteristics of caspase-3, -6, -7, and -8. J Biol Chem 272:25719–25723
Lanneau D, Brunet M, Frisan E, Solary E, Fontenay M, Garrido C (2008) Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med 12:1–19
Mjahed H, Girondon F, Fontenay M, Garrido C (2012) Heat shock proteins in hematopoietic malignancies. Exp Cell Res 318:1946–1958
Jego G, Hazoume R, Seigneuric C, Garrido C (2013) Targeting heat shock proteins in cancer. Cancer Lett 332:275–285
Kennedy D, Jäger R, Mosser DD, Samali A (2014) Regulation of apoptosis by heat shock proteins. IUBMB Life 65:327–338
Garrido C, Bruey JM, Fromentin A, Hammann A, Arrigo AP, Solary E (1999) HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J 13:2061–2070
Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G (2006) Heat shock proteins 27 and 70: antiapoptotic proteins with tumorigenic properties. Cell Cycle 5:2592–2601
Mosser DD, Morimoto RI (2004) Molecular chaperones and the stress of oncogenesis. Oncogene 23:2907–2918
Aghdassi A, Phillips P, Dudeja V, Dhaulakhandi D, Sharif R, Dawra R, Lerch MM, Saluja A (2007) Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res 67:616–625
Choi DH, Ha JS, Lee WH, Song JK, Kim GY, Park JH, Cha HJ, Lee BJ, Park JW (2007) Heat shock protein 27 is associated with irinotecan resistance in human colorectal cancer cells. FEBS Lett 581:1649–1656
Luo X, Tao L, Lin P, Mo X, Chen H (2012) Extracellular heat shock protein 72 protects Schwann cells from hydrogen peroxide-induced apoptosis. J Neurosci Res 90:1261–1269
Taylor AR, Robinson MB, Gifondorwa DJ, Tytell M, Milligan CE (2007) Regulation of heat shock protein 70 release in astrocytes: role of signaling kinase. Dev Neurobiol 67:1815–1829
Bejarano I, Espino J, Marchena AM, Barriga C, Paredes SD, Rodríguez AB, Pariente JA (2011) Melatonin enhances hydrogen peroxide-induced apoptosis in human promyelocytic leukaemia HL-60 cells. Mol Cell Biochem 353:167–176
Bejarano I, Redondo PC, Espino J, Rosado JA, Paredes SD, Barriga C, Reiter RJ, Pariente JA, Rodríguez AB (2009) Melatonin induces mitochondrial-mediated apoptosis in human myeloid HL-60 cells. J Pineal Res 46:392–400
Espino J, Bejarano I, Paredes SD, Barriga C, Reiter RJ, Pariente JA, Rodríguez AB (2011) Melatonin is able to delay endoplasmic reticulum stress-induced apoptosis in leukocytes from elderly humans. Age 33:497–507
Espino J, Bejarano I, Paredes SD, Barriga C, Rodríguez AB, Pariente JA (2011) Protective effect of melatonin against human leukocyte apoptosis induced by intracellular calcium overload: relation with its antioxidant actions. J Pineal Res 51:195–206
Samali A, Orrenius S (1998) Heat shock proteins: regulators of stress response and apoptosis. Cell Stress Chaperones 3:228–236
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK (2002) Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 277:15028–15034
Vabulas RM, Ahmad-Nejad P, Ghose S, Kirchning CJ, Issel RD, Wagner H (2002) HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem 277:15107–15112
Svensson PA, Asea A, Englund MC, Bausero MA, Jernas M, Wiklund O, Ohlsson BG, Carlsson LM, Carlsson B (2006) Major role of HSP70 as a paracrine inducer of cytokine production in human oxidized LDL treated macrophages. Atherosclerosis 185:32–38
Tidwell JL, Houenou LJ, Tytell M (2004) Administration of HSP70 in vivo inhibits motor and sensory neuron degeneration. Cell Stress Chaperones 9:88–98
Guzhova I, Kislyakova K, Moskaliova O, Fridlanskaya I, Tytell M, Cheetham M, Margulis B (2001) In vitro studies show that Hsp70 can be released by glia and that exogenous Hsp70 can enhance neuronal stress tolerance. Brain Res 914:66–73
Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS (2004) Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 287:817–833
Kahya MC, Naziroglu M, Cig B (2014) Seleium reduces mobile phone (900 MHz)-induced oxidative stress, mitochondrial function, and apoptosis in breast cancer cells. Bio Trace Elem Res 160:285–293
Naziroglu MC, Tokat S, Demirci S (2012) Role of melatonin on electromagnetic radiation-induced oxidative stress and Ca2+ signaling molecular pathways in breast cancer. J Recept Signal Transduct Res 36:290–297
Barbouti A, Doulias PT, Nousis L, Tenopoulou M, Galaris D (2002) DNA damage and apoptosis in hydrogen peroxide-exposed Jurkat cells: bolus addition vs. Continuous generation of H2O2. Free Radic Biol Med 33:691–702
Yu Y, Liu M, Zhang L, Cao Q, Zhang P, Jiang H, Zou Y, Ge J (2012) Heat shock transcription factor 1 inhibits H2O2-induced cardiomyocyte death through suppression of high-mobility group 1. Mol Cell Biochem 364:263–269
Kiang JG, Ding XZ, McClain DE (1996) Thermotolerance attenuates heat-induced increases in [Ca2+]i and HSP-72 synthesis but not heat-induced intracellular acidification in human A-431 cells. J Investig Med 44:53–63
Acknowledgments
This work was supported by Gobierno de Extremadura grant IB13072. J. Espino holds a research post-doctoral fellowship from Gobierno de Extremadura ((jointly financed by the European Regional Development Fund (ERDF); ref. PO1401).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Additional information
Lourdes Franco and Jorge Terrinca have contributed equally to this study.
Rights and permissions
About this article

Cite this article
Franco, L., Terrinca, J., Rodríguez, A.B. et al. Extracellular heat shock proteins protect U937 cells from H2O2-induced apoptotic cell death. Mol Cell Biochem 412, 19–26 (2016). https://doi.org/10.1007/s11010-015-2604-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-015-2604-y