
Abstract
Ischemic heart disease is the leading cause of death for both men and women worldwide, accruing 7.4 million deaths in 2012. There has been a continued search for better cardioprotective modalities that would reduce myocardial ischemia–reperfusion injury. Among these attempts, a more convenient model of ischemic preconditioning, known as remote ischemic preconditioning (RIPC) was first introduced in 1993 by Przyklenk and colleagues who reported that brief regional occlusion–reperfusion episodes in one vascular bed of the heart render protection to remote myocardial tissue. Subsequently, major advances in myocardial RIPC came with the use of skeletal muscle as the ischemic stimulus. To date, numerous studies have revealed that RIPC applied to the kidney, liver, mesentery, and skeletal muscle, have all exhibited cardioprotective effects. The main purpose of this review article is to summarize the new advances in understanding the molecular mechanisms of RIPC during the past 5 years, including those related to capsaicin-activated C sensory fibers, hypoxia-inducible factor 1α, connexin 43, extracellular vesicles, microRNA-144, microRNA-1, and nitrite. In addition, we have discussed results from several recent human clinical trials with RIPC. Taken together, the emerging clinical evidence supports the concept that the effectiveness of RIPC paired with its low-cost and non-invasive features makes it an ideal treatment before reperfusion after sustained ischemia. More carefully designed studies are warranted to fully exploit the clinical benefits of RIPC and its potential implications in patients with cardiovascular disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic heart disease (IHD) is the leading cause of death for both men and women worldwide, accruing 7.4 million deaths in 2012 (http://www.who.int/mediacentre/factsheets/fs310/en/). The detrimental effects of acute ischemia–reperfusion injury on myocardial tissue continue to plague IHD patients. Myocardial infarct size is a major determining factor of prognosis and current approaches to improve survival are directed at achieving early myocardial reperfusion with fibrinolytic therapy or via percutaneous coronary intervention. Paradoxically, even early revascularization leads to tissue damage, a phenomenon known as ischemia-reperfusion injury (IRI). This prompted a search for cardioprotective mechanisms that would reduce the infarct size in acute settings and protect against IRI.
In 1986, Murry et al. discovered that multiple, brief nonlethal ischemic episodes precondition the heart and reduce infarct size caused by a subsequent ischemic insult, a phenomenon known as myocardial ischemic preconditioning (IPC) [1]. Subsequent studies showed that IPC also decreases the incidence and severity of post-ischemic arrhythmias and enhances the recovery of cardiac function after global ischemia, attenuating the risk of ischemic reperfusion injury [2, 3].
Przyklenk and Whittaker in 1993, extended the concept of IPC by showing that brief regional occlusion–reperfusion episodes in one vascular bed of the heart render protection to remote myocardial tissue [4]. Since then, researchers began looking into more convenient models of preconditioning, known as remote ischemic preconditioning (RIPC). A major advance in myocardial RIPC came with the use of skeletal muscle as the ischemic stimulus. Kharbanda et al. produced RIPC with tourniquet occlusion of blood flow to one hind limb of an experimental pig model in 2002 [5]. To date, studies have revealed that preconditioning applied to the kidney, liver, mesentery, and skeletal muscle have all exhibited cardioprotective effects [6] (Table 1).
Detailed understanding of the cardioprotective pathways induced by RIPC has been thoroughly discussed by Hausenloy and Yellon [7] in 2008 and Costa et al. [8] in 2013. The present review provides update on the molecular mechanisms responsible for myocardial RIPC and its use in the clinical setting.
Potential mechanisms of remote ischemic preconditioning
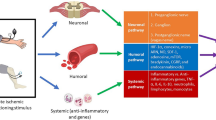
While preconditioning has proven valuable in reducing IRI, it has provided unprecedented opportunity to search for novel pharmacological interventions, which trigger similar molecular pathways as RIPC. The actual mechanism through which RIPC works is currently unclear but can be divided into three main components: (1) humoral, (2) neural, and (3) systemic factors of cardioprotection.
Humoral factors in RIPC
The humoral hypothesis posits that the ischemic stimulus leads to the production of substances that enter the circulation and reach the myocardium, where they exert a protective effect. This hypothesis was consolidated by Dickson et al. in 1999 who reported that blood taken from a rabbit that underwent simultaneous IPC of both heart and kidney, could reduce myocardial infarct size by 77 % when transfused into an isolated rabbit [9].
In 2009, Shimizu et al. identified several important facts related to the humoral mechanism in which they prepared dialysate of remotely preconditioned rabbit and human hearts [10]. The plasma dialysate derived from humans during RIPC conferred protection in rabbits, suggesting that the humoral substance(s) had cross-species effects. Further, they confirmed the presence of transferable hydrophobic <15 kDa cardioprotective factor(s) in plasma from remotely preconditioned rabbits, which demonstrated resistance to freezing, thawing, and denaturing. However, the actual identity of the humoral mediator remains unknown. Other studies have investigated whether endogenous substances such as adenosine [11], bradykinin [12], opioids [13], calcitonin gene-related peptide (CGRP) [14], and endocannabinoids [15], are generated in the remote tissues. It is proposed that these RIPC mediators enter the bloodstream and activate their respective receptors in the myocardium, thereby recruiting various intracellular pathways of cardioprotection. However, the humoral factors responsible for RIPC still remain unclear and further studies are required.
Neural factors of RIPC
The neural hypothesis postulates that substances produced in the remote ischemic territory act locally via afferent neural pathways, activating various efferent pathways that induce cardioprotection. In support of this notion, hexamethonium, a ganglion blocker, abolished the cardioprotective effects elicited by RIPC [16]. The theory was further developed with the proposition that endogenous substances such as adenosine [11], bradykinin [12], CGRP [14], released by the remote preconditioned organ, stimulated afferent nerve fibers, which then relay to efferent nerve fibers terminating on the myocardium to confer cardioprotection.
Ding et al. demonstrated that renal nerve section abolished the cardioprotective effect induced by renal ischemia (preconditioning) stimulus providing supportive evidence of a neural pathway [17]. Further confirmatory evidence implicating adenosine in a neural pathway of cardioprotection was provided by Liem et al. [18]. These findings suggested that adenosine released locally from the mesenteric bed during IRI stimulated the mesenteric afferent sensory nerves, which helps activate myocardial adenosine receptors. Dong et al. demonstrated that dissecting the femoral nerve abolished the myocardial infarct-limiting effect of remote hind limb preconditioning, suggesting that an intact neural pathway is required for the sensory afferent neural signaling from the preconditioning limb [19].
Schoemaker and van Heijningen reported that bradykinin administration was able to reduce myocardial infarct size, but was sensitive to hexamethonium [12]. Furthermore, the cardioprotective effect was abolished when HOE-140, a bradykinin B2 specific receptor antagonist, was administered prior to brief mesenteric artery occlusion–reperfusion. Similar to the studies on adenosine, these results indicate that bradykinin generation, through mesenteric RIPC, may stimulate afferent nerves, which then activate cardioprotective mechanisms in myocardial tissue. Wolfrum et al. not only confirmed these results but also showed that PKC-ϵ activation was blocked by HOE-140 and hexamethonium, individually [20]. They later studied CGRP, a neurotransmitter released from capsaicin-sensitive sensory nerves, as a potential mediator of RIPC [21]. Remote intestinal preconditioning generates nitric oxide which stimulates capsaicin-sensitive sensory nerves in the intestinal vasculature, releasing CGRP into the bloodstream then carried to the heart where it activates myocardial PKC-ε [22, 23].
An interesting study by Jones et al. further elucidated a neurogenic role to cardioprotection via capsaicin, PKC-ε, and KATP signaling. Instead of IPC, these authors studied the effects of “nonischemic” preconditioning by surgically producing an abdominal slit in mice in order to activate sensory nerves under the skin [24]. It was observed that skin nociception activated cardiac sensory and sympathetic nerves to elicit cardioprotective effects. Bradykinin, a known hormone and neurotransmitter, is released from sympathetic nerves in the heart and triggers a cascade that ultimately activated PKC-ε and inactivated PKC-δ in cardiomyocytes. In addition, topical application of capsaicin-activated C sensory fibers in the skin which significantly reduced IRI.
Systemic factors
In 2004, Konstantinov et al. sought to determine the effects of RIPC in inflammatory gene transcription in humans. Using microarray technology, they found that within 15 min of preconditioning, genes encoding proteins involved in cytokine synthesis, leukocyte chemotaxis, adhesion and migration, exocytosis, innate immunity, signaling pathways, and apoptosis were all suppressed—even more so after 24 h. Leukocyte CD11b expression also decreased significantly after 24 h, showing that RIPC suppressed pro-inflammatory gene transcription in human leukocytes, helping to confer the protective role of RIPC against IRI [25]. However, Albrecht et al. presented some contradictory results, showing that serum cytokines were actually elevated within the first phase of RIPC [26]. It is expected that during reperfusion, both pro- and anti-inflammatory molecules will act together to restore organ function while preventing tissue damage. A recent study showed an improvement in effective healing and cardioprotection due to the increase in the number of neutrophils just after bypass in the right atrial tissue [26]. This was also corroborated by an increase in the levels of IL-8, IL-1β, and TNF-α—major inflammatory regulators—in the RIPC group versus the control group.
Molecular mechanisms
Based on experimental studies, it has been widely understood that mechanisms underlying RIPC mirror that of classic IPC and postconditioning. These include binding of ligands to G-protein cell surface coupled receptors such as adenosine [11], bradykinin [12], opioids [13], angiotensin [27], and endocannaboids [15], nitric oxide (NO), activation of intracellular kinases such as PKC-ϵ [20], transcription factors, generation of reactive oxygen species (ROS) [28], and opening mitochondrial KATP channel [11]. These mechanisms are reviewed as follows.
ATP-sensitive potassium channel (KATP)
In IPC studies, KATP channels have demonstrated to be a key trigger in conferring cardioprotection. Within the IPC model, it is proposed that the signal transduction cascade terminates at the mitochondria, causing opening of the mitochondrial KATP channels. This, in turn, generates ROS, which can mediate cardioprotection by up regulating pro-survival kinases or inhibiting the opening of the mitochondrial permeability transition pore (mPTP) [29]. mPTP is a high-conductance channel of the inner mitochondrial membrane, whose opening in the first few minutes of myocardial reperfusion mediates cell death through ATP depletion and mitochondrial swelling [30]. Specifically in RIPC, Konstantinov et al. showed that the cardioprotective effects in myocardial tissue act through a mitochondrial KATP channel pathway [31].
Nuclear factor kappa B (NFκB)
Diwan et al. investigated the potential role of NFκB in erythropoietin-mediated cardioprotection by employing a selective NFκB inhibitor [32]. They concluded that erythropoietin preconditioning and remote renal RIPC triggered similar signaling mechanisms for activation, i.e., NFκB activation followed by opening of mitochondrial KATP channels.
Nitric oxide (NO) and nitrite
NO has been implicated as a major mediator of cardioprotection in IPC [29]. However, the potential role of NO in RIPC cardioprotection has yielded opposing results, i.e., the protective effect of RIPC was not abolished by NO inhibition [33]. Xiao et al. reported that intestinal ischemia upregulated CGRP levels, but administration of an nitric oxide synthase inhibitor abolished RIPC-induced cardioprotection [23]. These authors suggested that the delayed cardioprotection yielded by CGRP occurs via NO-dependent pathway. While there is a strong likelihood for other factors to influence the level of CGRP and other compensatory mechanisms, further studies are needed to elucidate the role of NO in the RIPC cardioprotection paradigm.
When the heart is subjected to ischemia, nitrite is reduced by deoxymyoglobin to form NO in the cardiomyocyte, limiting cellular injury and infarction [34, 35]. Rassaf et al. in 2014 reported that circulating nitrite derived from shear stress-dependent stimulation of endothelial nitric oxide synthase (eNOS) at the remote site of remote IPC contributed to cardioprotection during IRI. Interestingly, pharmacological and genetic inhibition of NO and nitrite generation by eNOS at the remote site or nitrite bioactivation by myoglobin within the target organ abrogated the cardioprotection by RIPC. Transfer experiments of plasma from healthy volunteers subjected to RIPC identified plasma nitrite as a cardioprotective agent in isolated Langendorff mouse heart preparations exposed to IRI [35].
Protein kinase C
PKC is a well-known mediator of cardioprotection [29]. The activation of PKC in the heart by CGRP, adenosine, and bradykinin B2, appears to be one of the earliest events in the myocardial mechanisms of cardioprotection [29]. The IPC cardioprotective mechanism is mediated by kinases including PI3 kinase, ERK/MAPK, PKC, and JAK/STAT [36]. These kinases prevent opening of mPTP, preservation of ATP thereby facilitating mitochondrial and myocardial protection. IPC also activates pro-survival kinases, resulting in the inhibition of the mPTP, but whether RIPC also activates these pro-survival kinases is unclear [36]. Heidbreder et al. demonstrated that mitogen-activated protein kinases (MAPKs) were activated within the intestines, but not within the cardiac tissue following intestinal ischemia–reperfusion [37]. It was not clear if the MAPKs were activated later as part of the RISK pathway during reperfusion.
New emerging mechanisms
Hypoxia-inducible factor 1α (HIF-1α)
Albrecht et al. recently demonstrated the involvement of HIF-1α in RIPC-induced cardioprotection in 32 patients undergoing cardiopulmonary bypass [26]. During four 5-min cycles of transient upper limb ischemia–reperfusion, HIF-1α accumulation and activation began in right atrial tissue. This was associated with reduced activities of caspases 3 and 7 (two markers for cell apoptosis) and significantly reduced serum levels of troponin T (a marker for cardiomyocyte necrosis) in the RIPC patients as compared with 29 control patients during the first 48-h postoperative period. While a causative link between increased HIF-1α levels and cardioprotection by RIPC is not conclusive, it is likely that the induction and/or stabilization of HIF-1α in the heart may be triggered by RIPC-mediated hypoxia-like events and the release of humoral factors, such as several cytokines—IL-8, IL-1β, and TNFα, that eventually reach remote regions including the right atria via blood circulation in these RIPC patients [26]. Various studies have corroborated that HIF-mediated signaling in cardiac tissue regulates myocardial damage, apoptosis, and inflammation, whereas HIF-1α has also been proposed to play a central role in cardioprotection by IPC [38–40].
Connexin 43 (Cx43)
Cx43 is an integral membrane protein that is mainly localized on sarcolemma of cardiomyocytes where six connexin molecules assemble into a connexon or hemichannel. Brandenburger et al. has demonstrated that Cx43 is critically involved in cardioprotective interventions including IPC [41]. These authors investigated the influence of RIPC on the expression patterns of Cx43 after IRI in the rat heart in vivo. IRI caused a strong decrease of relative Cx43 protein expression in the area at risk that was partly abolished by RIPC. Furthermore, RIPC decreased the level of ischemia-induced dephosphorylation of Cx43. Confocal immunofluorescence staining showed that I/R caused a loss of the Cx43 signal at the intercalated disks, while the Cx43 signal at the intercalated disks was partly sustained after RIPC. Thus, preservation of Cx43 protein expression and phosphorylation after RIPC might have a protective role.
Extracellular vesicles
Extracellular vesicles are membrane-bound structures, which contain a high concentration of RNAs and proteins. Since they can be secreted and specifically taken up by other cells, they are prime medium for intercellular signal transfer mechanisms. Giricz et al. demonstrated release of extracellular vesicles from the heart IPC stimuli is increased and these vesicles were responsible for the transmission of remote conditioning signals for cardioprotection [42]. Further cellular and molecular mechanistic studies are warranted to decipher the nature of actual effector factors carried by these vesicles.
Micro RNA-144
In 2014, Li et al. demonstrated a role of microRNA 144 (miR-144) in RIPC-induced cardioprotection. Using microRNA microarray, these authors showed that RIPC increased miR-144, whereas IRI decreased levels in the mouse heart [43]. Moreover, IRI was attenuated by both RIPC and intravenous administration of miR-144 in these studies. The systemic treatment with miR-144 increased phosphorylation of several kinases, i.e., Akt, GSK3β, and p44/42 MAPK. In addition, there was decrease in the phosphorylated mTOR levels and enhanced autophagy signaling. Importantly, systemic administration of a specific antisense oligonucleotide reduced myocardial levels of miR-144 and abrogated cardioprotection by RIPC. These authors also showed that RIPC increased plasma levels of miR-144 in mice and humans. While there was no change in plasma micro-particle (50–400 nM) numbers or their miR-144 content, a ~4-fold increase in miR-144 precursor in the exosome pellet and a significant increase in miR-144 levels was observed in the exosome-poor serum with associated increase in the miR carriage protein-argonaute-2 levels [43]. These data suggested that systemic release of microRNA 144 plays a pivotal role in the cardioprotection induced by RIPC and plasma miR-144 could potentially serve as a biomarker of the effectiveness of RIPC.
Micro RNA-1
Brandenburger et al. examined the involvement of microRNA 1 (miR-1) in RIPC [44]. In these studies, ischemia alone had no effect on miR-1 expression, whereas RIPC led to a downregulation of miR-1 prior to ischemia as well as after 2 h of reperfusion. However, after 6 h of reperfusion, RIPC caused increase in miR-1. Furthermore, luciferase assays confirmed the interaction of miR-1 with brain-derived neurotrophic factor (BDNF), a protein that exerts cardioprotective effects. However, miR-1 levels did not correlate with protein levels of BDNF, a known target of miR-1 in vivo [44]. The biological significance of changes in miR-1 expression levels and the potential interaction with BDNF in RIPC-induced cardioprotection needs further investigations.
Overall, much of the mechanistic studies in RIPC are still a work in progress. Further studies are needed to establish the direct cause and effect relationship of the various active molecules involved in the cardioprotective effect of RIPC. In such a complex pathway with various factors, it is likely that neither the humoral nor the neural pathways are mutually exclusive.
Translational studies in humans
RIPC has gone from experimental studies with animals to proof of principle studies in humans. The value of RIPC has been evaluated in populations ranging from pediatric patients undergoing cardiac surgery to adult patients undergoing elective abdominal aortic aneurysm repair, coronary angioplasty, and coronary artery bypass surgery [45]. Cardiac surgery is strongly associated with IRI, which leads to myocardial necrosis and mortality. Most, but not all, of these studies showed attenuation in the release of cardiac enzymes in RIPC-treated cohorts versus matched controls [45]. The first clinical trial with RIPC was conducted in 37 pediatric patients who underwent cardiopulmonary bypass during repair of congenital heart defects [46]. Compared to the 20 control group children, 17 RIPC group children that were subjected to four 5-min cycles of lower limb ischemia and reperfusion presented with much lower troponin levels, suggesting reduced damage to the heart postoperatively (Fig. 1). Levels of IL-10 were significantly increased 3 h postoperatively in the RIPC group, while TNFα was significantly decreased in this group (Fig. 2). In another landmark study by Hausenloy et al., 57 patients who underwent coronary artery bypass graft (CABG), there was a 43 % decrease in the serum troponin T levels in the RIPC group, indicating reduced myocardial damage [47]. To study its safety and efficacy, Thielmann et al. demonstrated that RIPC provided perioperative myocardial protection and improved prognosis in this CABG patient population [48]. In patients undergoing elective percutaneous coronary intervention (PCI), similar results were found: troponin T levels and ischemia-induced chest discomfort were reduced in the RIPC group versus control group [49].

Pre- and post-operative levels of troponin I in remote ischemic preconditioning (RIPC) and control groups. Adopted with permission from Cheung et al. [46]

Levels of interleukin 10 (IL-10) at 3 h and tumor necrosis factor-α (TNF-α) at 6 h postoperatively in remote ischemic preconditioning (RIPC) and control groups. Adopted with permission from Cheung et al. [46]
Similar to delayed or late phase of IPC, RIPC-induced cardioprotection has been shown to be a biphasic phenomenon, with an early first phase that lasts for up to 3 h after initial ischemia, followed by a delayed second phase that begins after 12–24 h, and lasts for up to 4 days [50]. A study by Loukogeorgakis et al. demonstrated that RIPC in humans offers up to 48 h of protection from myocardial reperfusion injury [50]. The most convenient aspect of this phenomenon is that RIPC could be triggered 24 h before cardiopulmonary bypass surgery, angioplasty, or transplantation while providing up to 48 h of resistance to cardiac ischemia–reperfusion injury.
Slagsvold et al., found that after coronary artery bypass surgery, only 14 % of RIPC patients developed atrial fibrillation during the first 3 days versus 50 % in the control group [51]. Interestingly, these authors did not observe a difference in the plasma concentrations of cardiac troponin T or creatine kinase between the RIPC group versus the control. Thus it appears that RIPC induces protection of the human atrium even without the increase in the cardiac markers of injury [50].
Most recently, Yellon’s group assessed the effect of RIPC on perioperative myocardial injury (PMI) in 180 patients undergoing elective CABG [52]. They reported that RIPC significantly reduced (1) magnitude of PMI by 26 %, (2) postoperative events of atrial fibrillation by over 50 %, and (3) incidence of acute kidney injury by 48 %. Interestingly, while intravenous glyceryl trinitrate (GTN), a donor of NO and vasodilator, decreased the incidence of PMI in the control group, RIPC was not beneficial to the patients who were administered GTN.
While the use of RIPC may seem irrelevant and impractical for more sudden ischemic episodes such as suffering a ST Segment Elevation Myocardial Infarction (STEMI), an interesting study by Bøtker et al. highlighted its potential merits [53]. Adult STEMI patients, while being transported to the hospital for PCI, were randomized to receive four 5-min cycles of ischemia–reperfusion. The results demonstrated that RIPC before hospital admission increased myocardial salvage compared to the control, and had a favorable safety profile.
Despite these encouraging results, two recent trials reported negative results on RIPC [54, 55]. McCrindle et al. showed that RIPC did not significantly improve clinical outcomes in pediatric patients undergoing cardiopulmonary bypass [54], possibly due to confounding factors such as the use of propofol during anesthesia, which is known to block the effectiveness of RIPC. Kono et al. explored the capability of RIPC to improve coronary microcirculation among healthy subjects and heart failure patients [55]. After 1-week course of RIPC, left ventricular ejection fraction was decreased among heart failure patients and microcirculation improved among healthy subjects without adverse effects. However, no long-term benefits of RIPC were observed in these patients. One of the limitation for this study was its small sample size (n = 20).
In summary, the effectiveness of RIPC paired with its low-cost and non-invasive features makes it an ideal treatment before reperfusion after sustained ischemia. Ovize et al. noted that 13 out of the 25 published phase II trials, showed statistically significant positive cardioprotection, 5 showed positive protection (significance not achieved), and 7 demonstrated no benefit or worsened myocardial injury [6]. As such, some enthusiasm has been lost for the effectiveness of RIPC. However, it is important to note that in these studies, the patient population may have contributed to the discrepancy in results. For example, some studies exclusively enrolled stable patients undergoing CABG, whereas others enrolled high-risk patients, who may have had multiple surgeries, which clearly heightens the risk for poor outcomes. Furthermore, the anesthesia regimen is widely varied from study to study, thereby confounding the results and distracting from the potentially invaluable clinical benefit of RIPC. Therefore, more carefully designed studies are warranted to fully explore the clinical benefits of RIPC and its potential implications in patients with cardiovascular disease.
References
Murry CE, Jennings RB, Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124–1136
Hagar JM, Hale SL, Kloner RA (1991) Effect of preconditioning ischemia on reperfusion arrhythmias after coronary artery occlusion and reperfusion in the rat. Circ Res 68:61–68
Lott FD, Guo P, Toombs CF (1996) Reduction in infarct size by ischemic preconditioning persists in a chronic rat model of myocardial ischemia-reperfusion injury. Pharmacology 52:113–118
Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P (1993) Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 87:893–899
Kharbanda RK, Mortensen UM, White PA, Kristiansen SB, Schmidt MR, Hoschtitzky JA, Vogel M, Sorensen K, Redington AN, MacAllister R (2002) Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation 106:2881–2883
Ovize M, Thibault H, Przyklenk K (2013) Myocardial conditioning: opportunities for clinical translation. Circ Res 113:439–450
Hausenloy DJ, Yellon DM (2008) Remote ischaemic preconditioning: underlying mechanisms and clinical application. Cardiovasc Res 79:377–386
Costa JF, Fontes-Carvalho R, Leite-Moreira AF (2013) Myocardial remote ischemic preconditioning: from pathophysiology to clinical application. Rev Port Cardiol 32:893–904
Dickson EW, Reinhardt CP, Renzi FP, Becker RC, Porcaro WA, Heard SO (1999) Ischemic preconditioning may be transferable via whole blood transfusion: preliminary evidence. J Thromb Thrombolysis 8:123–129
Shimizu M, Tropak M, Diaz RJ, Suto F, Surendra H, Kuzmin E, Li J, Gross G, Wilson GJ, Callahan J, Redington AN (2009) Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: evidence suggesting cross-species protection. Clin Sci (Lond) 117:191–200
Pell TJ, Baxter GF, Yellon DM, Drew GM (1998) Renal ischemia preconditions myocardium: role of adenosine receptors and ATP-sensitive potassium channels. Am J Physiol 275:H1542–H1547
Schoemaker RG, van Heijningen CL (2000) Bradykinin mediates cardiac preconditioning at a distance. Am J Physiol Heart Circ Physiol 278:H1571–H1576
Patel HH, Moore J, Hsu AK, Gross GJ (2002) Cardioprotection at a distance: mesenteric artery occlusion protects the myocardium via an opioid sensitive mechanism. J Mol Cell Cardiol 34:1317–1323
Tang ZL, Dai W, Li YJ, Deng HW (1999) Involvement of capsaicin-sensitive sensory nerves in early and delayed cardioprotection induced by a brief ischaemia of the small intestine. Naunyn Schmiedebergs Arch Pharmacol 359:243–247
Hajrasouliha AR, Tavakoli S, Ghasemi M, Jabehdar-Maralani P, Sadeghipour H, Ebrahimi F, Dehpour AR (2008) Endogenous cannabinoids contribute to remote ischemic preconditioning via cannabinoid CB2 receptors in the rat heart. Eur J Pharmacol 579:246–252
Gho BC, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD (1996) Myocardial protection by brief ischemia in noncardiac tissue. Circulation 94:2193–2200
Ding YF, Zhang MM, He RR (2001) Role of renal nerve in cardioprotection provided by renal ischemic preconditioning in anesthetized rabbits. Sheng Li Xue Bao 53:7–12
Liem DA, Verdouw PD, Ploeg H, Kazim S, Duncker DJ (2002) Sites of action of adenosine in interorgan preconditioning of the heart. Am J Physiol Heart Circ Physiol 283:H29–H37
Dong JH, Liu YX, Ji ES, He RR (2004) Limb ischemic preconditioning reduces infarct size following myocardial ischemia-reperfusion in rats. Sheng Li Xue Bao 56:41–46
Wolfrum S, Schneider K, Heidbreder M, Nienstedt J, Dominiak P, Dendorfer A (2002) Remote preconditioning protects the heart by activating myocardial PKCepsilon-isoform. Cardiovasc Res 55:583–589
Wolfrum S, Nienstedt J, Heidbreder M, Schneider K, Dominiak P, Dendorfer A (2005) Calcitonin gene related peptide mediates cardioprotection by remote preconditioning. Regul Pept 127:217–224
Li YJ, Xiao ZS, Peng CF, Deng HW (1996) Calcitonin gene-related peptide-induced preconditioning protects against ischemia-reperfusion injury in isolated rat hearts. Eur J Pharmacol 311:163–167
Xiao L, Lu R, Hu CP, Deng HW, Li YJ (2001) Delayed cardioprotection by intestinal preconditioning is mediated by calcitonin gene-related peptide. Eur J Pharmacol 427:131–135
Jones WK, Fan GC, Liao S, Zhang JM, Wang Y, Weintraub NL, Kranias EG, Schultz JE, Lorenz J, Ren X (2009) Peripheral nociception associated with surgical incision elicits remote nonischemic cardioprotection via neurogenic activation of protein kinase C signaling. Circulation 120:S1–S9
Konstantinov IE, Arab S, Kharbanda RK, Li J, Cheung MM, Cherepanov V, Downey GP, Liu PP, Cukerman E, Coles JG, Redington AN (2004) The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol Genomics 19:143–150
Albrecht M, Zitta K, Bein B, Wennemuth G, Broch O, Renner J, Schuett T, Lauer F, Maahs D, Hummitzsch L, Cremer J, Zacharowski K, Meybohm P (2013) Remote ischemic preconditioning regulates HIF-1alpha levels, apoptosis and inflammation in heart tissue of cardiosurgical patients: a pilot experimental study. Basic Res Cardiol 108:314
Singh D, Chopra K (2004) Evidence of the role of angiotensin AT1 receptors in remote renal preconditioning of myocardium. Methods Find Exp Clin Pharmacol 26:117–122
Weinbrenner C, Nelles M, Herzog N, Sarvary L, Strasser RH (2002) Remote preconditioning by infrarenal occlusion of the aorta protects the heart from infarction: a newly identified non-neuronal but PKC-dependent pathway. Cardiovasc Res 55:590–601
Yellon DM, Downey JM (2003) Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev 83:1113–1151
Hausenloy DJ (2013) Cardioprotection techniques: preconditioning, postconditioning and remote conditioning (basic science). Curr Pharm Des 19:4544–4563
Konstantinov IE, Li J, Cheung MM, Shimizu M, Stokoe J, Kharbanda RK, Redington AN (2005) Remote ischemic preconditioning of the recipient reduces myocardial ischemia-reperfusion injury of the denervated donor heart via a KATP channel-dependent mechanism. Transplantation 79:1691–1695
Diwan V, Kant R, Jaggi AS, Singh N, Singh D (2008) Signal mechanism activated by erythropoietin preconditioning and remote renal preconditioning-induced cardioprotection. Mol Cell Biochem 315:195–201
Petrishchev NN, Vlasov TD, Sipovsky VG, Kurapeev DI, Galagudza MM (2001) Does nitric oxide generation contribute to the mechanism of remote ischemic preconditioning? Pathophysiology 7:271–274
Corti P, Gladwin MT (2014) Is nitrite the circulating endocrine effector of remote ischemic preconditioning? Circ Res 114:1554–1557
Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M (2014) Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res 114:1601–1610
Hausenloy DJ, Lecour S, Yellon DM (2011) Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxid Redox Signal 14:893–907
Heidbreder M, Naumann A, Tempel K, Dominiak P, Dendorfer A (2008) Remote vs. ischaemic preconditioning: the differential role of mitogen-activated protein kinase pathways. Cardiovasc Res 78:108–115
Charron CE, Chou PC, Coutts DJ, Kumar V, To M, Akashi K, Pinhu L, Griffiths M, Adcock IM, Barnes PJ, Ito K (2009) Hypoxia-inducible factor 1alpha induces corticosteroid-insensitive inflammation via reduction of histone deacetylase-2 transcription. J Biol Chem 284:36047–36054
Eckle T, Kohler D, Lehmann R, El KK, Eltzschig HK (2008) Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation 118:166–175
Loor G, Schumacker PT (2008) Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ 15:686–690
Brandenburger T, Huhn R, Galas A, Pannen BH, Keitel V, Barthel F, Bauer I, Heinen A (2014) Remote ischemic preconditioning preserves Connexin 43 phosphorylation in the rat heart in vivo. J Transl Med 12:228
Giricz Z, Varga ZV, Baranyai T, Sipos P, Paloczi K, Kittel A, Buzas EI, Ferdinandy P (2014) Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J Mol Cell Cardiol 68:75–78
Li J, Rohailla S, Gelber N, Rutka J, Sabah N, Gladstone RA, Wei C, Hu P, Kharbanda RK, Redington AN (2014) MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res Cardiol 109:423
Brandenburger T, Grievink H, Heinen N, Barthel F, Huhn R, Stachuletz F, Kohns M, Pannen B, Bauer I (2014) Effects of remote ischemic preconditioning and myocardial ischemia on microRNA-1 expression in the rat heart in vivo. Shock 42:234–238
Przyklenk K, Whittaker P (2011) Remote ischemic preconditioning: current knowledge, unresolved questions, and future priorities. J Cardiovasc Pharmacol Ther 16:255–259
Cheung MM, Kharbanda RK, Konstantinov IE, Shimizu M, Frndova H, Li J, Holtby HM, Cox PN, Smallhorn JF, Van Arsdell GS, Redington AN (2006) Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery: first clinical application in humans. J Am Coll Cardiol 47:2277–2282
Hausenloy DJ, Mwamure PK, Venugopal V, Harris J, Barnard M, Grundy E, Ashley E, Vichare S, Di SC, Kolvekar S, Hayward M, Keogh B, MacAllister RJ, Yellon DM (2007) Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomised controlled trial. Lancet 370:575–579
Thielmann M, Kottenberg E, Kleinbongard P, Wendt D, Gedik N, Pasa S, Price V, Tsagakis K, Neuhauser M, Peters J, Jakob H, Heusch G (2013) Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: a single-centre randomised, double-blind, controlled trial. Lancet 382:597–604
Hoole SP, Heck PM, Sharples L, Khan SN, Duehmke R, Densem CG, Clarke SC, Shapiro LM, Schofield PM, O’Sullivan M, Dutka DP (2009) Cardiac Remote Ischemic Preconditioning in Coronary Stenting (CRISP Stent) Study: a prospective, randomized control trial. Circulation 119:820–827
Loukogeorgakis SP, Panagiotidou AT, Broadhead MW, Donald A, Deanfield JE, MacAllister RJ (2005) Remote ischemic preconditioning provides early and late protection against endothelial ischemia-reperfusion injury in humans: role of the autonomic nervous system. J Am Coll Cardiol 46:450–456
Slagsvold KH, Rognmo O, Hoydal M, Wisloff U, Wahba A (2014) Remote ischemic preconditioning preserves mitochondrial function and influences myocardial microRNA expression in atrial myocardium during coronary bypass surgery. Circ Res 114:851–859
Candilio L, Malik A, Ariti C, Barnard M, Di SC, Lawrence D, Hayward M, Yap J, Roberts N, Sheikh A, Kolvekar S, Hausenloy DJ, Yellon DM (2014) Effect of remote ischaemic preconditioning on clinical outcomes in patients undergoing cardiac bypass surgery: a randomised controlled clinical trial. Heart. doi:10.1136/heartjnl-2014-306178
Botker HE, Kharbanda R, Schmidt MR, Bottcher M, Kaltoft AK, Terkelsen CJ, Munk K, Andersen NH, Hansen TM, Trautner S, Lassen JF, Christiansen EH, Krusell LR, Kristensen SD, Thuesen L, Nielsen SS, Rehling M, Sorensen HT, Redington AN, Nielsen TT (2010) Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet 375:727–734
McCrindle BW, Clarizia NA, Khaikin S, Holtby HM, Manlhiot C, Schwartz SM, Caldarone CA, Coles JG, Van Arsdell GS, Scherer SW, Redington AN (2014) Remote ischemic preconditioning in children undergoing cardiac surgery with cardiopulmonary bypass: a single-center double-blinded randomized trial. J Am Heart Assoc. doi:10.1161/JAHA.114.000964
Kono Y, Fukuda S, Hanatani A, Nakanishi K, Otsuka K, Taguchi H, Shimada K (2014) Remote ischemic conditioning improves coronary microcirculation in healthy subjects and patients with heart failure. Drug Des Dev Ther 8:1175–1181
Ding YF, Zhang MM, He RR (2000) Ischemic preconditioning reduces cardiomyocytic apoptosis in rabbit heart in vivo. Sheng Li Xue Bao 52:220–224
Zhang SZ, Wang NF, Xu J, Gao Q, Lin GH, Bruce IC, Xia Q (2006) Kappa-opioid receptors mediate cardioprotection by remote preconditioning. Anesthesiology 105:550–556
Davidson SM, Selvaraj P, He D, Boi-Doku C, Yellon RL, Vicencio JM, Yellon DM (2013) Remote ischaemic preconditioning involves signalling through the SDF-1alpha/CXCR4 signalling axis. Basic Res Cardiol 108:377
Acknowledgments
This study was supported in part by Grants from National Institutes of Health (HL51045, HL79424, HL118808 to RCK) and the American Heart Association (14GRNT20010003 to FNS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Rabia Gill and Robin Kuriakose have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Gill, R., Kuriakose, R., Gertz, Z.M. et al. Remote ischemic preconditioning for myocardial protection: update on mechanisms and clinical relevance. Mol Cell Biochem 402, 41–49 (2015). https://doi.org/10.1007/s11010-014-2312-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-014-2312-z