
Abstract
Caveolin-1 (Cav-1), a protein component of cellular membrane, has been reported to regulate several cancer cell behaviors. However, its role on cancer metastasis in anoikis resistant cells is unknown. The present study aimed to investigate the correlation between Cav-1 level and aggressive behaviors of anoikis resistant cancer cells. Cav-1 and ShRNACav-1 stably transfected lung carcinoma cells, and anoikis resistant H_AR1 and H_AR2 cells expressing different levels of Cav-1 were subjected to anoikis, cell growth, anchorage-independent growth, extracellular matrix adhesion, cisplatin sensitivity, migration, and invasion assays. The correlations between cellular Cav-1 level and such cancer aggressive behaviors were evaluated. Results revealed that anoikis resistant lung cancer cells as well as Cav-1 overexpressing cells exhibit a significant increase in anchorage-independent growth, extracellular matrix adhesion, migration, and invasion in comparison to those of their parental H460 cells. Knock-down of Cav-1 by ShRNA transfection was able to reverse such metastatic potentials in H_AR2 cells. In addition, basal Cav-1 level of these cells was positively correlated with anoikis resistance, anchorage-independent growth, migration, and invasion behaviors of the cells, whereas such Cav-1 level showed poor correlation to cisplatin sensitivity, cell adhesion, and growth in attached condition. These findings give more information regarding role of Cav-1 in the regulation of behaviors of lung cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the leading cause of cancer death worldwide [1, 2]. Although therapeutic techniques and chemotherapies for the treatment of lung cancer are much developed nowadays, the outcome remains poor with only less than 15 % of patients surviving in the first 5 years [3]. Not only do lung cancer cells exhibit high rate of metastasis, but also the aggressiveness of their secondary tumors attenuates the patients’ survival, and is considered as major causes of poor prognosis and difficulty in managing the disease [4]. Metastasis is a multistep biological process, and the regulation of metastasis is extremely complicated [5]. The early step of metastasis requires cancer cells to detach and migrate away from the primary tumor, and to intravasate into blood or lymphatic circulations. This detachment in the most cases leads to cellular processes of apoptosis called anoikis (detachment-induced apoptosis) [6]. Defects in anoikis lead to the survival of cells during their transportation through the circulation system, and reach distant target organs, where they adhere, extravasate, and begin to form secondary tumors [4, 7].
Recently, many attempts have been made to investigate the mechanism(s) in controlling secondary cancer aggressiveness found in these anoikis resistant cancer populations [8–10]. The cancer cell aggressiveness has been generally described as the rate of secondary tumor growth, invasiveness, and chemotherapeutic resistance [8]. Many phenotypic changes have been suspected to contribute to lung cancer cell aggressiveness involving up-regulation of oncogenes and down-regulation of tumor suppressor genes [11–13]. Caveolin-1 (Cav-1), the protein that has been shown to implicate in anoikis resistance, has garnered increase attention since their regulatory roles in several cellular behaviors were continuously demonstrated [14–16]. Indeed, Cav-1, a 21–24 kDa structural protein component of the plasma membrane microdomains termed caveolae, has been shown to function in vesicular trafficking, signal transduction, and cancer progression [17–19]. Although up-regulation of this protein normally occurs in a variety of terminally differentiated cells including fibroblast, adipocytes, smooth muscle cells, endothelial cells, and epithelial cells [20], Cav-1 is greatly reduced in most oncogenically transformed and cancer cells [21–23]. Thus, Cav-1 was first explained to function as a tumor suppressor protein [24, 25]. In contrast, growing evidence indicates its role as a tumor and metastatic promoter as overexpression or re-expression of Cav-1 was found in many advanced stages and metastatic cancer cells. Up-regulation of Cav-1 was shown to render Rat1A cells more resistant to apoptosis [26]. Moreover, antisense-induced down-regulation of Cav-1 caused human prostate cancer cells more sensitize to apoptosis [27]. Previous studies have shown that Cav-1 acts as a negative regulator of anoikis [14, 15, 28, 29], and its elevated expression in lung carcinoma is closely associated with the increased metastasis capacity and poor survival of patients [16].
Anoikis resistance can be driven in originally sensitive clones by altering culture cycles under adhered and non-adhered (suspended media) conditions [30]. The effect of anoikis acquisition on aggressive behaviors as well as metastatic potentials of lung cancer cells is largely unknown. Besides, while cancer behaviors like growth, invasion, and chemotherapeutic resistance are considered as hallmarks of advanced lung cancer, the role of Cav-1 protein in regulation of mentioned cell behaviors is still elusive. Using pharmacological, genetic manipulation, and correlation analysis approaches, we have demonstrated an important role of Cav-1 protein and anoikis acquisition on lung cancer cells behaviors. Our findings would broaden the knowledge which facilitates the development of new strategies for metastasis therapeutic interventions, and give remarkable insight into the treatment for cancer metastases.
Materials and methods
Cell culture
Non-small cell lung carcinoma H460 cells were obtained from American type culture collection (Rockville, MD). The cells were cultured and maintained in RPMI medium, supplemented with 10 % fetal bovine serum (GIBCO, USA), 2 mmol/l l-glutamine (GIBCO, USA) and 100 units/ml penicillin/streptomycin at 37 °C in 5 % carbon dioxide environment.
Selection for anoikis-resistant cell line
Anoikis-resistant cells were generated by sequential cycles of culturing under adhered and non-adhered conditions. Briefly, H460 cells monolayer was trypsinized, and a single-cell suspension was seeded onto polyHEMA-coated tissue culture plates for 72 h. After incubation, cells were washed and transferred to tissue culture plates and left to recover. After the surviving clones were expanded, the cells were seeded again in polyHEMA-treated dish as mentioned above, and the cycle was repeated for five times. The resulted phenotype cells then were selected for single cell colony designated as H_AR1 and H_AR2. The cells were maintained in adhered condition and passaged several times before being used in this study.
Reagents
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt (XTT) and agarose (low melting point) were obtained from Sigma Chemical, Inc. (St. Louis, MO, USA). Propidium iodide (PI) and Hoechst 33342 were obtained from Molecular Probes, Inc. (Eugene, OR, USA). PromoFectin transfecting agent was obtained from PromoKine (Heidelberg, Germany). Rabbit polyclonal anti-Cav-1 antibody and horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G antibody were purchased from Abcam (Cambridge, MA, USA). Primary antibody beta-actin and Cav-1 shRNA plasmid were purchased from Santa Cruz Biotechnology. Cav-1 (pEX_Cav-1-YFP) plasmid was obtained from American type culture collection (Rockville, MD). PcDNA3 plasmid was generously provided by Dr. Yon Rojanasakul (West Virginia University, Morgantown, WV, USA). Other biochemical reagents were from Sigma (St, Louis, MO, USA).
Stable Cav-1 overexpressed and Cav-1 knock-down cells generation
Cav-1 overexpressed H460 (H_Cav1-1 and H_Cav1-2) cells were generated by stable transfection of H460 cells with Cav-1 plasmid (pEX_Cav-1-YFP) from the American type culture collection (ATCC, USA). Cav-1 knock-down H460 (H_shCav1) cells were generated by stable transfection of H460 cells with Cav-1 shRNA plasmid (sc-29241-SH) from Santa Cruz Biotechnology, Inc. Stable transfections were performed using Promofectin® reagent according to manufacturer’s instruction. Briefly, H460 cells were cultured on 24-well plates until they reached 50–60 % confluence. One microgram of Cav-1 or Cav-1 shRNA plasmid and 2 µl of Promofectin solution were mixed and incubated at room temperature for 30 min, then added to the cells in the absence of serum. After 24 h, the medium was replaced with culture medium containing 10 % FBS. After appropriate selection, 200µl of 100 cells/ml of each transfectant was seeded into each well of 96-well cell culture plate and cultured until cell colony was clearly observed. A single colony in wells was picked up and further cultured for greater cell number. After propagation period, all clones were identified by western blotting analysis and the clones that exhibit two different expression levels comparing to endogenous Cav-1 level of H460 cells were indicated as H_Cav1-1 and H_Cav1-2 cells. For the cells transfected with Cav-1 shRNA, the clones that stably express at least 50 % reduction of Cav-1 level compared to H460 cells were considered as H_shCav1 cells.
Anoikis assay
Cells were trypsinized and seeded at the number of 1 × 104 cells onto polyHema-Coated ninety-six-well tissue culture plates. After incubated for indicated time, cell viability was determined by incubation with 0.3 mg/ml of XTT at 37 °C for 4 h. The intensity of formazan product was determined at 450 nm using an ELISA plate reader. Relative cell viability was calculated by dividing the OD reading of the sample by that of the control.
Cell viability assay
Cells were trypsinized and seeded at the number of 1 × 104 cells onto ninety-six-well tissue culture plates. After treatment and incubated for indicated time, cell viability was determined by incubation with 0.5 mg/ml of MTT at 37 °C for 4 h. The intensity of formazan product was determined at 550 nm using an ELISA plate reader. Relative cell viability was calculated by dividing the OD reading of the sample by that of the control.
Extracellular matrix (ECM) adhesion assay
The cells with density of 3 × 104 cells/well were plated into 96 well plates precoated with Matrigel and allowed to attach by incubation at 37 °C for 15 min. Non-adhered cells were removed by intense washing with PBS. Adherent cells were subsequently observed under a microscope,and the number of adhered cells per field was calculated in a number of random fields.
Cell morphology characteristic
The morphology of the non-confluent and confluent cells was characterized under microscope. Sulforhodamine B and phalloidin were used to stain cell cellular protein and filamentous actin, respectively. Sulforhodamine B-stained cells were examined under light microscope. Phallodin stained cells were examined under the fluorescent microscope.
Cell proliferation assay
1 × 104 cells were plated in 60 mm cell culture plate. The medium was changed every 2 days. Viable cells (trypan blue exclusion) were counted with Bio-Rad TC10™ automated cell counter from triplicate plates every other day up to 6 days. Cell proliferation was also evaluated by a colorimetric assay MTT-based. The assay was performed at 24, 48, 72, and 96 h after seeding. Briefly, 5 × 103 cells/well were seeded in 96 well plates. At indicated time, 0.5 mg/ml of MTT was added to the appropriate wells and continuous inoculation was performed for 4 h. The intensity of formazan product was determined at 550 nm using an ELISA plate reader. Relative cell viability was calculated by dividing the OD reading of the sample by that of the control.
Soft agar assay
A single-cell suspension was plated onto 24-well plate at density of 1 × 104 cells/well in 0.35 % low melting agarose over a 0.4 % agarose base layer culture medium. After 2-week incubation, the cells were stained with Hoechst 33342. Phase contrast images were recorded at day 0 and day 14. The colony number and length were evaluated by using GSA image analyzer software.
Transwell migration and invasion assays
2 × 105 cells in medium containing 1 % fetal bovine serum were seeded on the upper chamber of the transwell filter (3 µm pore). Medium containing 10 % fetal bovine serum was added to the lower chamber and incubated for 72 h at 37 °C . Following incubation, non-migrate cells were removed from the upper surface of the membrane by cotton wool. The cells attached to the lower chamber were fixed with methanol, stained with Hoechst 33342, and then counted under fluorescence microscope. For invasion assay, Matrigel (Becton–Dickinson, Bedford, MA, USA) was diluted in RPMI 1640 medium and 50 µl of the 0.5 mg Matrigel was evenly inoculated onto the upper chamber of a 6.5 mm transwell membrane and allowed to form a gel at 37 °C . Cells in medium containing 1 % fetal bovine serum were seeded on the upper chamber of the Matrigel-coated transwell filter (8 µm pore). Medium containing 10 % fetal bovine serum was added to the lower chamber and incubated for 72 h at 37 °C . Following incubation, non-invasive cells were removed from the upper surface of the membrane by cotton wool. The cells attached to the lower chamber were fixed with methanol, stained with Hoechst 33342, and then counted under fluorescence microscope.
Western Blotting assay
Cells were incubated with lyses buffer containing 2 % Triton X-100, 1 % sodium dodecyl sulfate (SDS), 100 mmol/L NaCl, 10 mmol/l Tris–HCl (pH 7.5), 1 mmol/L EDTA, and complete Mini cocktail protease inhibitor for 30 min on ice. After insoluble debris was pelleted by centrifuged at 14,000×g for 15 min at 4 °C, the supernatant was collected and determined for protein content using Bradford method. Proteins (40 µg) were resolved on 10 % SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto nitrocellulose membranes using standard procedures. The membranes were blocked in 5 % skim milk in TBST (25 mmol/l Tris–HCl, pH 7.4, 125 mmol/l NaCl, 0.1 % Tween 20) for 1 h, followed by incubation with appropriate primary antibodies at 4 °C overnight. Membranes were washed three times with TBST for 10 min, followed by incubation with horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature. The immune complexes were detected by chemiluminescence and quantified by imaging densitometer using analyst/PC densitometer software. Mean densitometry data from independent experiments were normalized with β-actin protein.
Statistical analysis
All the experiments were repeated at least three times. Statistical analysis was performed using one-way ANOVA. A p value of less than 0.05 would be considered as statistically significant.
Results
Anoikis resistant lung cancer cells exhibits enhanced growth in anchorage-independent condition
In order to study aggressive behaviors of anoikis resistant cells, we established anoikis-resistant sublines by subjecting lung carcinoma H460 cells to five sequential cycles of suspended and adhered culture as described [30]. Two single cells from anoikis resistant subline cells were collected and cultured under adhered condition, namely H_AR1 and H_AR2 cells. We characterized the aggressiveness of H_AR1 and H_AR2 cells by determining cell growth in normal culture and anchorage-independent conditions. The cells were analyzed for cell proliferation by cell counting and MTT viability assays at various time points. Figure 1a indicates the significantly higher number of viable cells in H_AR1 and H_AR2 groups than that of H460 control cells could be observed as early as day 4 in normal cultured condition. The result from MTT assay confirmed that the relative of cell viability was higher in H_AR1 and H_AR2 than that of parental H460 cells at 96 h after seeding.

Anoikis resistant lung cancer cells exhibits enhanced growth in attach and anchorage-independent conditions. a Viable H_AR1 and H_AR2 cells were counted with automated cell counter every other day up to 6 days. Values are mean ± SD (n = 6); * p < 0.05 versus control. b Viable cells were measured by MTT assay every 24 h up to 96 h. Values are mean ± SD (n = 6). * p < 0.05 versus control. c Single-cell suspensions were subjected to soft agar colony formation assay as described in materials and methods. Colonies were attained by light microscope and photographed after two weeks. d The relative cell colony diameter was determined by using image analyzer. Values are mean ± SD (n = 6); *p < 0.05 versus control. e The relative cell colony number was determined by using image analyzer. Values are mean ± SD (n = 6); *p < 0.05 versus control
Further, the ability of cells to grow in an anchorage-independent manner was evaluated by soft agar assay as described in materials and methods. The results indicated that acquisition of anoikis resistance significantly enhanced anchorage-independent growth of the cells as indicated by an increase of number of cell colony and colony size in the group of H_AR1 and H_AR2 cells in comparison to those of parental H460 cells (Fig. 1c–e).
Extracellular matrix (ECM) adhesion, migration, and invasion characteristics of H_AR1 and H_AR2 cells
Ability of cancer cells on ECM adhesion, migration, and invasion is considerated as an important factor in determining their metastasis potential. For ECM adhesion, H460, H_AR1, and H_AR2 cells were seeded into Matrigel-coated plates for 15 min, and the adhered cells were counted after the washing process. Figure 2a shows that H_AR1 and H_AR2 cells were more capable to adhere to ECM than the lung carcinoma H460 cells. The relative cell adhesion in H_AR1 and H_AR2 cells was ~0.15 and ~0.25 fold induction in comparison to that of control cells, respectively. Next, Boyden chamber migration assay was performed in order to determine migration ability of cells. The migrated cells in the lower chamber were stained with Hoechst 33342, and counted under fluorescence microscope. Figure 2b indicates that the acquisition of anoikis resistance in H_AR1 and H_AR2 cells enhanced migration of lung cancer cells. Compared with control cells, the relative cell migration of H_AR1 and H_AR2 cells was ~1 and ~2 fold, respectively. For invasion study, Boyden chamber invasion assay was used. The cells were seeded into Matrigel layer in upper chamber and incubated for 72 h. Invaded cells in lower chamber were stained with Hoechst 33342 and counted under fluorescence microscope. Figure 2c indicates that anoikis resistant subline cells exhibited higher degree of cell invasion over the control cells. Interestingly, these aggressiveness behaviors are corresponding with an increasing of Cav-1 expression (Fig. 2g).

Extracellular matrix (ECM) adhesion, migration, and invasion characteristics of H_AR1 and H_AR2 cells. a The cells were plated onto Matrigel-coated 96 well plates for 15 min. Adhered cells per field were calculated in a number of random fields. Values are mean ± SD (n = 6); *p < 0.05 versus control. b The cells were subjected into transwell migration assay. Migrated cells were stained with Hoechst 33342 and counted under fluorescence microscope. Values are mean ± SD (n = 6); *p < 0.05 versus control. c Invaded cells were stained with Hoechst 33342, and counted under fluorescence microscope. Values are mean ± SD (n = 6); *p < 0.05 versus control. d Cells were treated with various concentrations of cisplatin for 24 h and cell viability was determined by MTT assay. Values are mean ± SD (n = 6); *p < 0.05 versus control. e Sulforhodamine B-stained cells were examined under light microscope. f Phalloidin-stained cells were examined under fluorescent microscope. g Cav-1 expressions in H_AR1, H_AR2 and control cells were examined by Western blot analysis
Effect of anoikis resistance acquisition of the cells on cisplatin susceptibility
We tested whether acquisition of anoikis resistance of H460 cells plays a role in regulating cisplatin sensitivity. Cells were treated with various concentrations of cisplatin for 24 h, and cell viability was determined by MTT assay. The result showed that anoikis-resistant subline cells exhibited the same pattern of cisplatin-mediated cytotoxicity compared to H460 cells (Fig. 2d). Anoikis resistant subline cells were slightly more sensitive to cisplatin-mediated cytotoxic as compared to parental cells. This finding showed that the acquisition of anoikis resistance process in H460 cells has only a minimal impact on cisplatin susceptibility.
Acquisition of anoikis resistance alters cell morphology
To evaluate cell morphology and cytoskeletal structural organization, the cells were stained with sulforhodamine B and phalloidin. Sulforhodamine B and phalloidin were used to stain cellular protein and filamentous actin (F-actin), respectively. Acquisition of anoikis resistance increased cell spreading indicated by the increase of cell shape of H_AR1 and H_AR2 cells attached to cell culture plate (Fig. 2e). The staining of the cell with phalloidin demonstrated the increase of directional stress fiber and filopodia in anoikis-resistant subline cells (Fig. 2f).
Regulatory roles of Cav-1 on cancer aggressive behaviors
We and others have shown that Cav-1 plays a key a role in regulation of cancer metastasis including anoikis, migration, and invasion [28, 31–33]. However, the clear correlation between Cav-1 protein and cancer cell behaviors has not been demonstrated yet. In order to verify the mentioned correlations, we generated cells exhibiting several levels of Cav-1 by stable transfection with Cav-1 and shRNACav-1 plasmids. Then, the clone cells were evaluated for migration, invasion, cell growth, and growth in detached conditions as described. Figure 3a and b shows that overexpressed Cav-1 clone cells, H_Cav1-1 and H_Cav1-2, exhibited a significant increase of cell proliferation than that of H460 control cells, whereas the lowest cell proliferation capability was observed in Cav-1-knockdown cells in corresponding with Cav-1 expression (Fig. 3f). Also, the MTT-based proliferation assay was used to confirm the proliferative effect of Cav-1 (Fig. 3b). Further, the role of Cav-1 protein on cell growth in anchorage-independent condition was evaluated. Figure 3c-e indicates that the increase of Cav-1 in stable Cav-1 transfectants (H_Cav1-1 and H_Cav1-2 cells) resulted in enhancing growth of the cells in detached condition. This role was confirmed by the observation that a decrease in anchorage-independent growth was detected in Cav-1 knockdown H_shCav1 cells.

Effect of Cav-1 on cancer cell proliferation and anchorage-independent growth. a Viable H_Cav1-1, H_Cav1-2, and H460/shCav1 cells were counted every day up to 6 days. Values are mean ± SD (n = 6); * p < 0.05 versus control. b Viable cells were measure by XTT assay every 24 h up to 96 h. Values are mean ± SD (n = 6); * p < 0.05 versus control. c Single-cell suspensions were subjected to soft agar colony formation assay as described in materials and methods. Colonies were attained by light microscope and photographed after two weeks. d The relative cell colony diameter was determined by using image analyzer. Values are mean ± SD (n = 6); *p < 0.05 versus control. e The relative cell colony number was determined by using image analyzer. Values are mean ± SD (n = 6); *p < 0.05 versus control. f Cav-1 expressions in these transfectants were examined by Western blot analysis
Studies demonstrated regulatory effects of Cav-1 on ECM adhesion, migration, and invasion [16, 34, 35]. For ECM adhesion investigation, H460, H_Cav1-1, H_Cav1-2 cells, and H_shCav1 cells were seeded into Matrigel-coated plates for 15 min. After washed, adhered cells were counted. Figure 4a shows that Cav-1 over-expressed H460 cells exhibited higher ECM adhesion than the control H460 cells, while H_shCav1 cells exhibited less ability to adhere to the ECM. The role Cav-1 in migration ability of H460 cells was next evaluated by Boyden chamber migration assay. The results indicated that migration ability of Cav-1 overexpressed cells was increased, whereas such ability was reduced in H_shCav1 cells in comparison to that of control H460 cells. Boyden chamber invasion assay also revealed the same function of Cav-1 in enhancement of cancer cell invasion (Fig. 4c). The result of cisplatin-mediated cytotoxic assay indicated that Cav-1 manipulated cells exhibited only slightly alteration on cisplatin sensitivity (Fig. 4d). Also, sulforhodamine B staining indicated that Cav-1 overexpressed cells were observed as enlarged, elongated cells compared to the control cells (Fig. 4e). Meanwhile, rounded cells and retraction of cell–cell contact were observed in Cav-1-knock down H_shCav1 cells. The staining with phalloidin demonstrated that Cav-1 overexpressed cells had higher directional stress fiber, filopodia, and polarity as shown in Fig. 4f.

Effect of Cav-1 on cancer cell adhesion, migration, and invasion. a H_Cav1-1, H_Cav1-2, and H460/shCav1 cells were plated onto Matrigel-coated 96 well plates for 15 min. Adhered cells per field were calculated in a number of random fields. Values are mean ± SD (n = 6); *p < 0.05 versus control. b The cells were subjected into migration assay. Migrated cells were stained with Hoechst 33342, and then counted under fluorescence microscope. Values are mean ± SD (n = 6); *p < 0.05 versus control. c The cells were subjected into transwell invasion assay, invaded cells were stained with Hoechst 33342, and then counted under fluorescence microscope. Values are mean ± SD (n = 6); *p < 0.05 versus control. d Cells were treated with various concentrations of cisplatin for 24 h and cell viability was determined by XTT assay. Values are mean ± SD (n = 6). *p < 0.05 versus control. e Sulforhodamine B-stained cells were examined under light microscope. f Phalloidin-stained cells were examined under fluorescent microscope
Cav-1 is responsible for cancer cell aggressiveness in anoikis resistance driven cells
To provide supporting evidence that Cav-1 was responsible in the increased cell aggressiveness, we tested whether the decrease of Cav-1 in anoikis resistant cells can modulate the cells to be less aggressive. Anoikis-resistant H_AR2 cells were transfected with shRNA-Cav-1 plasmid and named as H_AR2/shCav1 cells. Western blotting analysis confirmed that Cav-1 level in H_AR2/shCav1 cells decreased in comparison to that of H_AR2 cells. The H_AR2/shCav1 cells were then analyzed for anoikis resistance, cell growth, anchorage-independent growth, ECM adhesion, migration, and invasion capacities. Figure 5 shows that H_AR2/shCav1 cells exhibited significantly decreased capability than those of H_AR2 cells in anoikis resistance and cell aggressiveness, suggesting that Cav-1 is responsible protein in regulation of cell aggressiveness in anoikis resistant cells.

Cav-1 up-regulation is responsible for cancer cell aggressiveness in anoikis-resistant driven cells a Anoikis-resistant H_AR2 cells were transfected with shRNA Cav-1 plasmid. Cav-1 level of the cells was determined by western blotting. b Cells were detached and suspended in poly-HEMA coated plates for the indicated times and cell viability was determined by XTT assay. Values are mean ± SD (n = 6); * p < 0.05 versus control. c Cells were investigated for cell growth, anchorage-independent number of colonies and colony length, adhesion, migration, and invasion as described in “Materials and methods.” Values are mean ± SD (n = 6); * p < 0.05 versus control
Evaluation for the correlations between Cav-1 level and aggressive behaviors of lung cancer cells
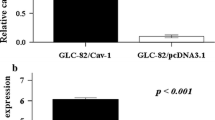
It is very interesting to verify the correlations between protein level and cell behaviors. The Cav-1 level of H460, H_AR1, H_AR2, H_Cav1-1, H_Cav1-2, and H_shCav1 cells was evaluated by western blotting and densitometry analysis. The data representing ability of H460, H_AR1, H_AR2, H_Cav1-1, H_Cav1-2, and H_shCav1 cells on anoikis resistance, cell growth, anchorage-independent growth, anchorage-independent survival, adhesion, migration, invasion and cisplatin resistance were collected and plotted against their basal Cav-1 levels. Correlation coefficient of each behavior was evaluated and is indicated in the Fig. 6 revealing that in these cells, the basal Cav-1 level correlated well with anoikis-resistant, anchorage-independent growth, cell migration, and cell invasion, while the protein level was less correlated with cell adhesion and cisplatin sensitivity as r value < 0.9.

Correlation analysis of basal Cav-1 level and cell aggressive behaviors. Correlation analysis between Cav-1 level and a relative anoikis resistance, b relative cell growth, c relative anchorage-independent colony length, d relative anchorage-independent colony number, e relative cell adhesion, f relative cell migration, g relative cell invasion and h relative cisplatin resistance was generated by Microsoft Excel 2007. Six independent experiments were performed. Correlation coefficient, r was determined from linear regression. i Cav-1 expressions in these transfectants were examined by Western blot analysis
Discussion
The ability of cancer cells escape from anoikis is a pre-requisite for cell metastasis by affording them to increase survival time while migrating to secondary sites. Thus, resistance to anoikis is considered as a crucial step during tumorigenesis. Acquisition of anoikis resistance in anoikis sensitive cancer cells can be driven by alternating culture cycles under adhered and suspended conditions. To date, acquisition of anoikis resistance has successfully been developed in many cells such as osteosarcoma, colorectal, hepatoma, and oral tongue cancer cells [30, 36–38]. Evidence has been piled up to suggest that acquisition of anoikis resistance can be selective pressure for cancer cell aggressiveness [36, 38–40]. In liver cancer, anoikis-resistant driven cells have more invasive capability, evading from cancer therapeutic agents and escaping from host immune attacks than anoikis-sensitive cells [40]. Furthermore, acquisition of anoikis resistance increased colorectal cancer cells tumorigenicity in vivo [36]. In addition, the study by Zhu et al. in melanoma cells reported that acquisition of anoikis resistance stimulated cell proliferation and invasiveness [39]. However, the effect and underlying mechanism of acquisition of anoikis resistance on cell aggressiveness in NSCLC are still unknown. In this study, we showed that when anoikis-susceptible lung carcinoma H460 cells were cultured under anoikis-inducing condition in vitro, they spontaneously gave rise to anoikis-resistant cells and manifesting dramatically increased aggressiveness. Cell growth, anchorage-independent growth, ECM adhesion, migration, and invasion were increased in H_AR1 and H_AR2 cells (Fig. 1 and 2). Interestingly, many cell aggressive behaviors were correspondently related to the level of anoikis resistance of the cells. These observations led to the hypothesis that the mechanism which cell has developed during anoikis acquisition may concomitantly increase aggressive potency of the cancer cells.
The processes of de-exposure to anoikis-inducing environment may act as a driving selection toward anoikis-resistant phenotype cells, or force adaptive anoikis resistant response of the cells [30]. Although the mechanisms of acquisition of anoikis resistance are largely unknown, many underlying mechanisms of cancer cells resisting to detachment-induced apoptosis have been proposed and the modification in oncogenes and tumor suppressor genes are by far the most mentioned [11, 13, 41]. Oncogenic versions of H-, K-, N-Ras, and Ras-related proteins TC21 and R-Ras, were shown to inhibit anoikis in RIE-1 rat intestinal epithelial cells [11]. The study by McFall et al. showed that Bcl-2 and cytokine response modifier A (crmA) suppressed anoikis by attenuating the Jun-N-Terminal Kinase (JNK) activation in canine kidney cells [13]. Furthermore, the stabilization of epidermal growth factor receptor (EGFR) was shown to suppress anoikis in mammary epithelial cells [41]. From epigenetic studies, the alteration in the patterns of gene expression due to gene silencing and reprogramming when exposed to anoikis microenvironment could be responsible, at least in part, in acquisition of anoikis resistant phenomena [42–44]. Moreover, the study by Kupferman et al. showed that the pattern of S100P, KLK6 and CTNNAL1 expression was altered in anoikis-resistance cell lines compared to the anoikis-sensitive cells [45].
Cav-1 protein has garnered an increasing attention in the cancer metastasis research and shown to play a crucial role on cancer progression and metastasis. Many studies have shown that Cav-1 reduced anoikis and increased cell aggressiveness in various cancer cells including prostate cancer cells, small cell lung cancer cells, melanoma cells, and hepatocellular carcinoma cells [33, 46–48]. Furthermore, Cav-1 levels are high in metastatic and multidrug resistance (MDR) human cancer cell lines [49, 50]. In breast cancer cells, Cav-1 inhibited anoikis by blocking p53 activation [15]. Cav-1 is responsible for induction of migration and invasion of head and neck squamous cell carcinoma through an oncogene miR-13a [32]. In non-small cell lung cancer (NSCLC), our previous studies and others proved that Cav-1 increased anoikis resistance, migration, and invasion of the cells [28, 29, 51]. In addition, Cav-1 was found higher in brain metastases NSCLC [52]. Even though Cav-1 role in anoikis resistance and cell aggressiveness in NSCLC has been reported, whether Cav-1 is responsible for anoikis exposure adaptation process resulting in acquisition of anoikis resistance and driven cell aggressiveness at the same time is still unknown. Our study for the first time showed that acquisition of anoikis resistance up-regulated Cav-1 exhibited by the higher level of Cav-1 in anoikis resistant cell lines. We also proved that Cav-1 is the key regulator in anoikis resistance and cell aggressiveness since the level of Cav-1 was correlated with the increase of anoikis resistance and cell aggressiveness. In clinical study, Cav-1 expression significantly correlated with drug resistance and a poor prognosis in advanced NSCLC patients treated with gemcitabine-based chemotherapy [49]. However, Cav-1 has a sensitizing effect on cisplatin-mediated death of lung cancer cells through superoxide anion-dependent mechanism [53]. The present study is consistent with the previous findings that sequential cycles of culturing under adhered and suspended condition are not altered sensitivity to chemotherapeutic agents of human osteosarcoma cell line [54].
Our finding suggested that Cav-1 up-regulation was acquired during repeated response to anoikis stimuli. Further, we provided information that such Cav-1 up-regulation was through an increase of Cav-1 gene expression. Although the mechanism(s) that regulates Cav-1 transcription in response to anoikis acquisition process is under investigation, some possible mechanisms have been proposed. Emergence of oncogenic K-ras mutations promoted by acquisition of anoikis resistance was shown to increase tumorigenicity in vivo in colorectal cancer cells [36]. Interestingly, K-ras-mutated cell lines showed an elevated expression of Cav-1 [55], implying the possibility that Cav-1 up-regulation in the present study might be caused by K-ras mutation.
In conclusion, we have demonstrated for the first time that Cav-1 protein was up-regulated in response to the acquisition of anoikis resistance and the up-regulation of Cav-1 played an important role in regulation of several cancer cell behaviors. We also provided the evidence revealing the correlation of Cav-1 level and cancer cell aggressiveness including cell growth, anchorage-independent growth, ECM adhesion, migration, and invasion.
Abbreviations
- Cav-1:
-
Caveolin-1
- XTT:
-
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt
- PBS:
-
Phosphate-buffered saline
- poly-HEMA:
-
poly-2-hydroxyethyl methacrylate
- ShRNA:
-
Short hairpin RNA
- TBST:
-
Tris-buffered saline with 0.1 % tween
- PI:
-
Propidium iodide
References
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55:74–108
American Cancer Society (2014) Cancer Facts & Figures, 2014. Atlanta, Ga: American Cancer Society
Jemal A, Murray T, Samuels A et al (2003) Cancer statistics. CA Cancer J Clin 53:5–26
Hanahan D, Weiberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Isaiah JF (2002) The organ microenvironment and cancer metastasis. Differentiation 70:450–498
Gilmore AP (2005) Anoikis. Cell Death Differ 12(2):1473–1477
Craig DS, Kika A, Aaron DS (2008) Anoikis resistance and tumor metastasis. Cancer Lett 272:177–185
Witte JS, Katrina AB, Goddard KAB et al (2000) Genomewide scan for prostate cancer-aggressiveness loci. Am J Hum Genet 67:92–99
Kassambara A, Klein B, Moreaux J (2009) MMSET is overexpressed in cancers: link with tumor aggressiveness. Biochem Biophy Res Comm 379:840–845
Moehler M, Fring C, Mueller A et al (2008) VEGF-D expression correlates with colorectal cancer aggressiveness and is downregulated by cetuximab. World J Gastroenterol 14:4156–4167
McFall A, Ulku A, Lambert QT et al (2001) Oncogenic Ras blocks anoikis by activation of a novel effector pathway independent of phosphatidylinositol 3-kinase. Mol Cell Biol 21:5488–5499
Rosen K, Rak J, Leung T et al (2000) Rocks: multifunctional kinase in cell behavior. J Cell Biol 149:447–456
Frisch SM, Vuori K, Kelaita D, Sicks S (1996) A role for Jun-N-terminal kinase in anoikis: suppression by bcl-2 and crmA. J Cell Biol 135:1377–1382
Fiucci G, Ravid D, Reich R, Liscovitch M (2002) Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene 21:2365–2375
Ravid D, Maor S, Werner R, Liscovitch M (2005) Caveolin-1 inhibits cell detachment-induced p53 activation and anoikis by upregulation of insulin-like growth factor-1 receptors and signaling. Oncogene 21:1338–1347
Ho CC, Huang PH, Huang HY et al (2002) Up-regulated caveolin-1 accentuates the metastasis capability of lung adenocarcinoma by inducing filopodia formation. Am J Pathol 161:1647–1656
Galbiati F, Razani B, Lisanti MP (2001) Emerging themes in lipid rafts and caveolae. Cell 106:403–411
Rothberg KG, Heuser JE, Donzell WC et al (1992) Caveolin, a protein component of caveolae membrane coats. Cell 68(4):673–682
Scherer PE, Lisanti MP, Baldini G et al (1994) Induction of caveolin-1 during adipogenesis and association of GLUT4 with caveolin-rich vesicles. J Cell Biol 127:1233–1243
Razani B, Engelman JA, Wang XB et al (2001) Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276:38121–38138
Engelman JA, Zhang XL, Galbiati F, Lisanti MP (1998) Chromosomal localization, genomic organization, and developmental expression of the murine caveolin gene family (Cav-1, -2, and -3) Cav-1 and Cav-2 genes map to a known tumor suppressor locus (6-A2/7q31). FEBS Lett 429:330–336
Lee SW, Reimer CL, Oh P, Campbell DB, Schnitzer JE (1998) Tumor cell growth inhibition by caveolin-1 re-expression in human breast cancer cells. Oncogene 16:1391–1397
Koleske AJ, Baltimore D, Lisanti MP (1995) Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci U.S.A 92:1381–1385
Campbell L, Hollins AJ, Al-Eid A et al (1999) Caveolin-1 expression and caveolae biogenesis during cell transdifferentiation in lung alveolar epithelial primary cultures. Biochem Biophys Res Common 262:744–751
Mikol DD, Hong HL, Cheng HL, Feldman EL (1999) Caveolin-1 expression in schwann cells. Glia 27:39–52
Timme TL, Goltsov A, Tahir S et al (2000) Caveolin-1 is regulated by c-myc and suppress c-myc-induced apoptosis. Oncogene 19:3256–3265
Li L, Ren CH, Tahir SA, Ren C, Thompson TC (2003) Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A. Mol Cell Biol 23:9389–9404
Rungtabnapa P, Nimmannit U, Halim H, Rojanasakul Y, Chanvorachote P (2011) Hydrogen peroxide inhibits non-small cell lung cancer cell anoikis through the inhibition of caveolin-1 degradation. Am J Physiol 300:235–245
Chanvorachote P, Nimmannit U, Lu Y et al (2009) Nitric oxide regulates lung carcinoma cell anoikis through inhibition of ubiquitin-proteasomal degradation of caveolin-1. J Biol Chem 284:28476–28484
Diaz-Montero CM, McIntyre BW (2003) Acquisition of anoikis resistance in human osteosarcoma cells. Eur J Cancer 39:2395–2402
Luanpitpong S, Talbott SJ, Rojanasakul Y et al (2010) Regulation of lung cancer cell migration and invasion by reactive oxygen species and caveolin-1. J Biol Chem 285:38832–38840
Nohata N, Hanazawa T, Kikkawa N et al (2011) Caveolin-1 mediates tumor cell migration and invasion and its regulation by miR-133a in head and neck squamous cell carcinoma. Int J Oncol 38:209–217
Yeh D, Chen C, Sun M et al (2009) Caveolin-1 is an important factor for the metastasis and proliferation of human small cell lung cancer NCI-H446 cell. Anat Rec 292:1584–1592
Beardsley A, Fang K, Mertz H et al (2005) Loss of caveolin-1 polarity impedes endothelial cell polarization and directional movement. J Biol Chem 280:3541–3547
Goetz TG, Minguet S, Navarro-Lerida I et al (2011) Biochemical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 146:148–163
Derouet M, Wu X, May L et al (2007) Acquisition of anoikis resistance promotes the emergence of oncogenic K-ras mutations in colorectal cancer cells and stimulates their tumorigenicity in vivo. Neoplasia 9:536–545
Zhang Z, Cao L, Li J et al (2008) Acquisition of anoikis resistance reveals a synoikis-like survival style in BEL7402 hepatoma cells. Cancer Lett 267:106–115
Swan EA, Jasser SA, Holsinger FC et al (2003) Acquisition of anoikis resistance is a critical step in the progression of oral tongue cancer. Oral Oncol 39:648–655
Zhu Z, Sanchez-Sweatman O, Huang X et al (2001) Anoikis and metastatic potential of cloudman S91 melanoma cells. Cancer Res 61:1707–1716
Cao L, Han L, Zhang Z et al (2009) Involvement of anoikis-resistance in the metastasis of hepatoma cells. Exp Cell Res 315:1148–1156
Schafer ZT (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461:109–113
Barcellos-Hoff MH (2001) It takes a tissue to make a tumor: epigenetics, cancer and microenvironment. J Mammary Gland Biol Neoplasia 6:212–221
Coomber BL, Yu JL, Fathers KE et al (2003) Angiogenesis and the role of epigenetics in metastasis. Clin Exp Metastasis 20:215–227
Belinsky SA (2002) Epigenetics is alive and growing. Trends Mol Med 8:53–54
Kupferman ME, Patel V, Sriuranpong V et al (2007) Molecular analysis of anoikis resistance in oral cavity squamous cell carcinoma. Oral Oncol 43:440–454
Karam JA, Lotan Y, Roehrborn CG et al (2007) Caveolin-1 overexpression is associated with aggressive prostate cancer recurrence. Prostate 67:614–622
Felicetti F, Parolini I, Bottero L et al (2009) Caveolin-1 tumor-promoting role in human melanoma. Int J Cancer 125:1514–1522
Zhang ZB, Cai L, Zheng SG, Xiong Y, Dong JH (2009) Overexpression of Caveolin-1 in hepatocellular carcinoma with metastasis and worse prognosis: correlation with vascular endothelial growth factor, microvessel density and unpaired artery. Pathol Oncol Res 3:495–502
Ho CC, Kuo SH, Huang PH et al (2008) Caveolin-1 expression is significantly associated with drug resistance and poor prognosis in advanced non-small cell lung cancer patients treated with gemcitabine based chemotherapy. Lung Cancer 59:105–110
Lavie Y, Fiucci G, Liscovitch M (1998) Up-regulation of caveolae and caveolar constituents in multidrug-resistant cancer cells. J Biol Chem 273:32380–32383
Sunaga N, Miyajima K, Suzuki M et al (2004) Different roles for caveolin-1 in the development of non-small cell lung cancer versus small cell lung cancer. Cancer Res 64:4277–4285
Cassoni P, Daniele L, Maldi E et al (2009) Caveolin-1 expression in lung carcinoma varies according to tumour histotype and is acquired de novo in brain metastases. Histopathology 55:20–27
Pongjit K, Chanvorachote P (2011) Caveolin-1 sensitizes cisplatin-induced lung cancer cell apoptosis via superoxide anion-dependent mechanism. Mol Cell Biochem 358:365–373
Diaz-Montero CM, McIntyre BW (2005) Acquisition of anoikis resistance in human osteosarcoma cells does not alter sensitivity to chemotherapeutic agents. BMC Cancer 5:39
Lee SH, Lee SJ, Chung JY et al (2009) p53 secreted by K-Ras-Snail pathway is endocytosed by K-Ras-mutated cells; Implication of target-specific drug delivery and early diagnostic marker. Oncogene 28:2005–2014
Acknowledgments
This research has been supported by the Ratchadaphiseksomphot Endowment Fund 2013 of Chulalongkorn University (CU-56-384-HR). The authors would like to thank Mr. Krich Rajprasit, a proofreader.
Conflicts of interest
The authors report no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chanvorachote, P., Pongrakhananon, V. & Halim, H. Caveolin-1 regulates metastatic behaviors of anoikis resistant lung cancer cells. Mol Cell Biochem 399, 291–302 (2015). https://doi.org/10.1007/s11010-014-2255-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-014-2255-4