
Abstract
Among all health-related issues, the rising concerns about drug resistance led to the look for alternative pharmaceutical drugs that are effective against both infectious and noninfectious diseases. Antimicrobial peptides (AMPs) are small molecular peptides that play a crucial role in the innate immunity of various organisms. They have amphiphilic structure and net positive charge, allowing them to interact with membranes and hydrophobic surfaces, showing strong broad-spectrum activity against different microorganisms, including bacteria, fungi, and viruses. They also exhibit other host-beneficial activities, including immunomodulation, anti-inflammatory, tissue regeneration, etc. AMPs exhibit antimicrobial activity through wide mechanisms of action, particularly by focusing on intracellular targets to inhibit the synthesis of nucleic acids and proteins. These wide ranges of mechanisms of action of AMPs have contributed to the slow development of resistance against microorganisms. The increasing pathogen resistance is a major global public health threat, and AMPs are constantly being explored and developed as another treatment for viral diseases such as HIV infection and (COVID-19). This review analyzes the potential of AMPs to combat antimicrobial resistance developed by several antimicrobial-resistant (AMR) microorganisms against existing antibiotics. This review focuses on the highlights of the sources, synthesis, mode, and mechanism of action, the evaluation of several benefits, and the outline of various hurdles. The review has also included the possible solution to the limitations associated with the clinical applications of AMPs, along with its future perspectives and development needed in drug discovery against AMR pathogens.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Living things have faced huge health challenges from the early Dark Age to the recent outbreak of the coronavirus (COVID-19) pandemic. Modern medicine’s failure to prevent, cure, and treat diseases and infections was the primary cause of the first Dark Ages. Antibiotics became available as options for treatment, and that’s when the golden age was declared. Unfortunately, this was unable to last for very long because multi-drug resistant (MDR) strains started to emerge (Luo et al. 2022). Multidrug-resistant (MDR) and pan-drug-resistant (PDR) bacteria have spread widely worldwide. They are responsible for rising morbidity and mortality rates and major societal costs (Friedman 2016; Rima et al. 2021). The inadequate frequency of new antibiotic classes being discovered and the rapid growth of resistance to novel antibiotics highlight the need for newer therapeutic alternatives to antibiotics, such as lysine-based small compounds, vaccines, anti-virulence agents, Phage treatment and antimicrobial peptides (Rima et al. 2021). According to the World Health Organization, Antimicrobial resistance (AMR) is a growing worldwide health issue that can make it difficult to treat bacterial infections in some situations. It is one of the top 10 worldwide public health hazards to humanity, and it is expected to kill over 10 million people each year by 2050 (O’Neill 2016b). Initiatives have been made worldwide to reduce the spread of AMR. As a result, in 2015, a global action plan on Antimicrobial Resistance (GAP) was developed to implement it. National action strategies are also further being developed to reduce the spread of AMR. Furthermore, the WHO analysis requests immediate action to avert an antimicrobial resistance disaster and emphasizes the significant discovery and development of novel antibiotics. Therefore, there is a requirement for new compounds that are active against pathogens, particularly those that cause nosocomial infections and are prone to multidrug resistance. Several different therapies have been offered to address this issue, amongst these antimicrobial peptides (AMPs) being one of the most promising, having lived in nature for millions of years, and there is little or no resistance development so far (Yu et al. 2018). This lack of/slow development of resistance against microorganisms may be related to the presence of several mechanisms of action of AMPs against bacteria as opposed to the set targets utilized by antibiotics. It makes them more attractive than antibiotics, which build resistance quickly (Rojas-Pirela et al. 2023). Furthermore, unlike other treatments, AMPs are less hazardous because they are broken down into amino acids. The main aim of this review is to determine where these AMPs stand now in controlling MDR bacterial infections. This review will analyze their potential to combat AMR, replace existing antibiotics, evaluate their benefits, and outline the hurdles faced by R&D.
Antimicrobial peptides, also known as AMPs, are small molecular peptides (usually less than 100 amino acid residues in length) that widely exist in nature (Moretta et al. 2021a). The sources of antimicrobial peptides are bacteria, fungi, plants, insects, amphibians, fish, birds, mammals, and the human body. AMPs are essential components of essentially all biological innate immunity and play a crucial role in resistance to the invasion of foreign pathogens. AMPs have amphiphilic structure and net positive charge. Due to this, there is a strong interaction with membranes and hydrophobic surfaces (Moretta et al. 2021a; Van Eijk et al. 2020) showing strong broad-spectrum activity against various microorganisms such as bacteria, fungi, and viruses. AMPs have antimicrobial activity by focusing on intracellular targets to inhibit the synthesis of nucleic acids and proteins and regulate host immune responses. A major global public health threat is increasing Pathogen resistance. Mechanisms of AMPs can be a good solution to address this problem of the increasing resistance of pathogenic microorganisms to antibiotics. AMPs are constantly being explored and developed as another treatment for viral diseases such as HIV infection and coronavirus disease 2019 (COVID-19).
Advancements in Peptide Drug Development
Timeline
The development of modern therapeutic medications can be broadly divided into four historical periods: the first dark age, the golden age, the second dark age, and the new era (Fig. 1). The period before the use of modern drugs like antibiotics is known as the ‘‘first dark age’’. This time is also known as the Pre-antibiotic era; this period had extremely high rates of morbidity and mortality due to common bacterial infections. The death penalty was due to diseases like pneumonia, typhoid and tuberculosis. Therefore, finding treatment and cure options was necessary, which was the main need at that time (Lewies et al. 2019; Luo et al. 2022).

Evolution of drug development: a historical timeline
The golden era. It is also known as the modern antibiotic era due to the discovery of antibiotics. Indeed, antibiotics have played astonishing roles in treating and controlling bacterial infectious diseases, ranging from simple to deadly infections. Since the discovery of penicillin by Alexander Fleming in 1928, antibiotics have revolutionized medicine and saved countless lives. The long quest from the discovery of penicillin by Alexander Fleming in 1928 to its clinical application more than a decade later provided invaluable lessons that have shaped the course of drug development. Subsequently, several important antibiotics were discovered and released to the market (Luo et al. 2022).
During the second dark era, also known as the post-antibiotic era, there was an increase in the emergence of microbial drug resistance. Antibiotics’ remarkable golden period could not last forever. 1948, the pandemic known as penicillin-resistant Staphylococcus aureus (PRSA) was first reported. Methicillin was subsequently developed and made available in 1959 to treat PRSA infections. However, methicillin-resistant S. aureus (MRSA) has been identified. The World Health Organisation (WHO) identified drug-resistant microorganisms as a worldwide issue in 2001. Drug resistance-related mortality might be responsible for up to 10 million annual deaths by 2050, according to a recent, concerning study (O’Neill 2016a, b). It has been found that one of the main issues related to public health is microbial drug resistance (Fjell et al. 2012). Thus, there is an immediate need for alternative treatment agents that are efficient against infections and drug-resistant strains. Remarkable advancements in science and technology indeed mark a new era in therapeutics. This includes the development of peptide drugs, recombinant DNA technology, personalized medicines, Gene Therapy, Immunotherapy, machine learning, and artificial intelligence (Lewies et al. 2019).
History of Antimicrobial Peptides
In 1939, Dubos discovered antimicrobial peptides (AMPs) by isolating an antimicrobial agent from a soil Bacillus strain (Fjell et al. 2012). This extract has been demonstrated to protect mice against pneumococcal infections (Singh et al. 2023). The following year, after fractionating the extract, Hotchkiss and Dubos identified an antimicrobial peptide (AMP) and named it gramicidin. Gramicidin was shown to be useful for the topical treatment of wounds and ulcers, but it showed toxicity after intraperitoneal application. (Luo et al. 2023a) Tyrocidine, a further AMP, was identified in 1941 and demonstrated efficacy against Gram-positive and Gram-negative bacteria. (Xuan et al. 2023) it, however, was harmful to human blood cells. Another AMP was also obtained from the plant Triticumaestivum in the same year. Later, purothionin name was given to that AMP, and it was found that it is effective against some pathogenic bacteria and fungi. In 1956, the first animal-derived AMP defensin was extracted from rabbit leukocytes. The next year, lactoferrin from cow milk and bombinin from epithelia were discovered. Simultaneously, it was demonstrated that AMPs are present in the lysosomes of human leukocytes.
Natural AMPs are found in both prokaryotes (such as bacteria) and eukaryotes (such as protozoa, fungi, plants, insects, and animals) (Conlon & Sonnevend 2010). The majority of AMPs are constantly produced by certain cells, whereas the formation of some AMPs can be induced. Using the silk moth as a model organism, Hultmark and colleagues have shown that vaccination with Enterobacter cloacae can generate P9A and P9B in hemolymph (Yi et al. 2014).
Sources of Antimicrobial Peptides
A large number of AMPs with different and specific amino acid sequences have been isolated from multiple diversified forms of living organisms, including various microorganisms, plants, and animals (Fig. 2). All these organisms biosynthesize AMPs for various specified purposes, mainly including the completion of the life cycle in viruses, competing nature in different bacteria, and the host’s defensive nature in higher organisms (Bin Hafeez et al. 2021; Huan et al. 2020). The rate of biosynthesis of these AMPs depends on the needs of the microorganisms. In many organisms, most AMPs are found to be continuously biosynthesized, while some AMPs’ biosynthesis is inducible, especially during the infection (Bahar & Ren 2013).

Diverse origins of antimicrobial peptides: highlighting key sources
The microbial sources of AMPs are viruses (bacteriophages), bacteria (Gram-positive and Gram-negative species), and several fungal species. Bacteriophages are known to complete their life cycle by infecting the bacteria. Bacteriophages inject their genetic material within the bacterial cells to produce their multiple progeny virions during infection. After their synthesis within the bacterial cell, they are released by causing cell lysis (Bin Hafeez et al. 2021). To accomplish this, bacteriophages are found to biosynthesize various bacterial membrane-lytic peptides such as endolysins (lysins), virion-associated peptidoglycan hydrolases (VAPGHs), and depolymerases, which aid in the successful accomplishment of the viral life cycle in the bacterial cell (Rahman et al. 2021). Endolysins have been found to act on the peptidoglycan layer of bacterial cell walls and aid in releasing progeny virions by bacterial cell lysis. VAPGHs are another form of membrane-lytic peptides encoded by the bacteriophage DNA that aids in the injection of the viral genetic material within the bacterial cell by hydrolyzing the peptidoglycan layer, resulting in the lysis and pores formation in the cell wall. The bacteriophage depolymerases tend to degrade the polysaccharide layer of the bacterial cell envelope, resulting in the lysis of biofilms. Many of these peptides, including endolysins like lysin Lys AB2, Ply V12, and PK34 (Peng et al. 2017); VAPGHs like several glycosidases, amidases, and endopeptidases (Latka et al. 2017); and depolymerases like K1, K11, K21, K25, K30, K35, K64, K69, KN4, and KN5 have been isolated and utilized as an alternative or in synergism with the traditional antibiotics (Bin Hafeez et al. 2021; Pan et al. 2017). Many of these viral peptides isolated have been shown to act against both Gram-positive and Gram-negative types of bacteria. Depolymerase peptides have also been mostly utilized due to their potential anti-biofilm activity (Bin Hafeez et al. 2021).
Most bacterial species compete with other bacterial species for their food and survivability. Some bacterial species compete by inhibiting the growth of different bacterial species in their vicinity by releasing AMPs. The first AMP isolated from the bacteria was Nisin, which was found to have potent antibacterial activity against many Gram-positive and Gram-negative species and has been currently utilized as a natural preservative in many food industries (Gharsallaoui et al. 2016). Various types of ribosomally and non-ribosomally synthesized AMPs have been isolated from many Gram-positive and Gram-negative bacterial species (Tajbakhsh et al. 2017). Bacteriocins are the main class of ribosomally-synthesized AMPs found in both bacterial species. In Gram-positive bacteria, bacteriocins are classified into four subclasses based on their structures: lantibiotics, non-lantibiotics, large-sized bacteriocins, and uniquely structured bacteriocins (Choi et al. 2023; Simons et al. 2020). Several important bacteriocins that have been isolated and studied in Gram-positive bacteria are nisin, lacticin, cytolysin, salivaricins, leucocin, enterocin, lactocin, plantaricin, thermophilin, acidocin, pediocin, gassericin, circularin, uberolysin, zoocin, lysostaphin, and heleveticin. In Gram-negative bacteria, bacteriocins are classified into colicins, colicin-like bacteriocins, microcins, and phage tail-like bacteriocins (Bin Hafeez et al. 2021). Bacteriocins found in E. coli are called colicins, and several important colicins that have been studied are colicin A, B, D, E1, E2, E3, E4, E5, E6, E7, E8, E8, M, and U (Cameron et al. 2019). Bacteriocins are found to have similar structures and functions as that of colicins but are synthesized by other Gram-negative species, except E. coli, especially including P. aeruginosa (process) and some Klebsiella species (plebians) are called colicin-like bacteriocins (Bin Hafeez et al. 2021). Bacteriocins found in Enterobacteriaceae species are called microcins, structurally small peptides with molecular weights less than 10 kDa. Several important microcins that have been isolated and studied are microcins B17, C7, D93, E492, H47, J25, L, and V (Bin Hafeez et al. 2021). Another type of bacteriocins found in several Gram-negative species with high molecular weight and structural similarity to the phage tail are phage-like bacteriocins and they are classified into R-type, similar to Myoviridae phage tails (R-pyocins) and F-type, similar to Siphoviridae phage tails (F-pyocins) (Bhattacharjee et al. 2022; Scholl 2017). Polymyxins and tridecaptins are important classes of non-ribosomal AMPs. Several important polymyxins that have been isolated and studied are polymyxins A, B, C, D, E, F, M, P, S, and T. Among them, polymyxins B and E have been commercialized and extensively utilized clinically against various multidrug-resistant Gram-negative species, including Enterobacteriaceae species, A. baumannii, P. aeruginosa, and S. maltophilia (Trimble et al. 2016; Velkov et al. 2019). Tridecaptins are another class of linear, non-ribosomally synthesized AMPs found in Paenibacillus species. Several important tridecaptins subclass that have been studied are tridecaptins A, B, C, G, and M (Ballantine et al. 2019; Bann et al. 2021). Fungal defensins are an important class of AMPs found in fungal species that play an important role in the fungal innate immune defense system. Plectasin was the first fungal defensin isolated from Pseudoplectania nigrella that has shown a potent antimicrobial activity against various Gram-positive bacterial species (Bin Hafeez et al. 2021). Other important fungal defensins that have been isolated and studied for their antimicrobial activity include triintsin from Trichophyton interdigitale (Shen et al. 2018), copsin from Coprinopsis cinerea (Essig et al. 2014), and bldesin from Blastomyces dermatitidis (Luo et al. 2019). Another important class of AMPs is peptaibol, which is found in soil fungi Trichoderma species. This class includes the AMPs with short amino acid sequences (about 5–10 amino acids) with a higher proportion of non-proteogenic amino acids, which mostly include α-aminoisobutyric acid (Aib) and an amino alcohol group at the C-terminal, hence the name peptaibol (Duclohier 2010). Some of the important peptaibol that have been studied extensively are alamethicin from T. viridea, trichogin GA IV and tricholongin B from T. longibrachiatum, and saturnisporin SA from T. Saturnisporum (Bin Hafeez et al. 2021; Ramachander Turaga 2020). Some other important classes of fungal AMPs include amatoxins and phallotoxins produced by Amanita phalloides mushrooms and dikaritins produced by Ascomycetes (Li et al. 2022).
Many AMPs are found in plants and to date, many of these AMPs have been isolated from roots, seeds, flowers, stems, and leaves of various plant species. AMPs play an important role in the plant’s defence system, especially the cysteine-rich AMPs, containing multiple disulphide bonds with resultant compact structure and higher stability of these AMPs (Campos et al. 2018; Tam et al. 2015). Depending on the sequence-based structural conformations and the arrangement of disulfide bonds between cysteine residues, plant AMPs have been classified into several families, which include thionins (e.g.: purothionin from wheat endosperm), defensins (e.g.: MsDef1 from Medicago sativa), lipid transfer proteins (e.g., AceAMP1 LTP from onion seeds), puroindolines (e.g.: PINA and PINB from wheat), snakins (e.g.: StSN1 and StSN2 from potato), cyclotides (e.g.: cycloviolacin O1 and O2 from Viola odorata), hevein-like peptides (e.g.: WjAMP1 from the leaves of Wasabia japonica L.), knottin-type peptides (e.g.: PAFP-S from Phytolacca americana), and smallest IB-AMPs from the seeds of Impatiens balsamina (Li et al. 2022; Nawrot et al. 2014).
Kingdom Animalia is the largest source of AMPs, as a wide variety of AMPs are found in all the species of this kingdom, contributing to the rapid defence system that tends to protect them from pathogenic microorganisms. Many AMPs have been isolated and studied from the vast majority of these species, including invertebrates and vertebrates (Bin Hafeez et al. 2021). Invertebrate sources of AMPs include insects and other arthropods, mollusks, and nematodes (Mylonakis et al. 2016). Invertebrate defensins are the most predominant class of AMPs found in invertebrates and they are found to be homologous with the vertebrate β-defensins (Shafee et al. 2017). Some other important classes of AMPs found in invertebrates are cecropins (first found in Hyalophora cecropia), crustins (first found in crustaceans), mellitin (first found in Apis mellifera), Acaloleptin (first found in Acalolepta luxuriosa), lycotoxin (first found in Lycosa carolinensis), Cupiennin (first found in Cupiennius salei), Tenicin (first found in Tenebrio molitor), Drosomycin and Drosocin (first found in Drosophila melanogaster), and sapecin (first found in Sarcophaga peregrina) (Bin Hafeez et al. 2021).
Vertebrate sources of AMPs include several aquatic organisms, amphibians, reptiles, birds, and mammals, including humans. Out of all marine organisms, fish is one of the main sources of AMPs and the important classes of AMPs found in fishes are cathelicidins, β-defensins, hepicidins, piscidins, and histone-derived peptides (Wang et al. 2023). Other aquatic sources of AMPs are Aspergillus fumigatus (BTMF9), Lumulus polyphemus/American horseshoe crab (Polyphemusin-1), Amphibalanus amphitrite (AaCrus1), and Olivacillaria hiatula (Li et al. 2022). In amphibians, the major sources of AMPs are frogs, which consist of vast varieties of AMPs. Several important genera of amphibians that contribute to this enormous variety of AMPs include Ayltes, Ascaphus, Bombina, Hoplobatrachus, Kassina, Leptodactylus, Litoria, Hylarana, Pseudis, Hypsiboas, Agalchnis, Pachymedusa, Phyllomedusa, Uperoleia, Crinia, Xenopus, Silurana, Hymenochirus, Amelops, Pelophylax, Limnonectes, Bufo, Ceratophrys, Lithobates, Odorrana, and Rana. Some of the important families of AMPs found in these genera of amphibians include alyteserin, ascaphins, bombinins, tigerinins, kassinatuerins, aureins, caerins, citropins, dahleins, maculatins, fallaxins, fallaxidins, brevinins, temporins, esculentin, guentherin, ranacyclins, ranatuerins,palustrins, pseudins, dermaseptin, dermatoxin, distinctin, phylloseptin, phylloxin, plasticin, SPYY, PGLa, CPF, XPF, uperins, signiferins, hymenochirins, amolopins, hainanenins, palustrins, jingdongins, odorranains, buforins, and nigrocins (Bin Hafeez et al. 2021; Xu & Lai 2015). Higher animals, including reptiles, birds, and mammals are also important sources of AMPs and the AMPs in these animals are mostly synthesized by the cells of specific systems, including the lymphatic system, respiratory system, gastrointestinal tract, genitourinary tract, and several sensory organs, contributing to the direct and indirect innate immune responses at these sensitive sites. The major families of AMPs found in these animals are cathelicidins and defensins (Ageitos et al. 2017). CATH, cathelicidin BF, β-Defensin, pelovaterin, and TEWP (turtle egg-white protein) are some important classes of cathelicidin and defensins families found in reptiles (Bin Hafeez et al. 2021a; van Hoek 2014). Some other important families of AMPs found in reptiles are waprin (e.g.: omwaprin found in Oxyuranus microlepidotus), proline-rich peptides (e.g.: waglerin found in Trimeresurus wagleri), and crotamine defensin-like toxin (e.g.: crotamine found in Snake Venom) (van Hoek 2014). Among avians, chickens are the main source of AMPs, and the chickens’ cathelicidins are called fowlicidins. Other cathelicidins and cathelicidins-like peptides found in avians are Cc-CATH from Coturnix coturnix, Cj-CATH from Coturnix japonica, Cl-CATH from Columba livia, dCATH from Anas platyrhynchos, Pc-CATH from Phasianus colchicus, and CATH from Meleagris gallopavo (Bin Hafeez et al. 2021). The important classes of cathelicidins that have been studied extensively in mammals are indolicidin (cathelicidin 4), protegrins, equine cathelicidins (eCATHs), and Bactenecins. LL-37 is the most prominently studied cathelicidin in humans, which has shown potent antimicrobial activity, along with its efficient anti-biofilm nature, against various Gram-positive, Gram-negative, and biofilm-synthesizing species (Bin Hafeez et al. 2021). All three classes of defensins, including α-, β-, and θ-defensins, are found in mammals and tend to have potent antimicrobial efficiency against various pathogenic species. α-and β-defensins are the predominant class of AMPs found in humans, whereas, they lack the expression of θ-defensins (Li et al. 2022).
Design and Synthesis of Antimicrobial Peptides
Extraction of AMPs from the above sources is an easy process, but this conventional technique of extraction was not favorable for the scale-up production of AMPs, as the yield of the directly extracted AMPs from their respective sources is found to be very low, resulting in the consumption of more time and expensive production. So, chemical synthesis and fermentation involving genetic engineering were considered favorable sources for obtaining the AMPs on larger scales (Lima et al. 2021; Vanzolini et al. 2022a). The chemical methods for peptide synthesis include solid-phase peptide synthesis (SPPS) and liquid-phase peptide synthesis (LPPS), among which SPPS is the most widely used technique because of its high reliability and efficiency in producing the peptides (Behrendt et al. 2016; Yeo et al. 2021). Though, certain limitations associated with the SPPS technique like low diffusion of solvents, leading to incomplete couplings and deprotections and resulting in the utilization of excess solvents for the accomplishment of proper diffusion and reaction in SPPS, are resolved by utilizing the LPPS technique, that has been associated with higher crude purity, less solvent consumption, and greater ease of scaling than SPPS (Yeo et al. 2021). However, chemical synthesis has also been associated with certain limitations, including technical processes, high cost of synthesis, tedious and hectic downstream processing of synthesized peptides, and environmental issues due to the extensive use of solvents in chemical synthesis (Luo et al. 2023b). Another approach of genetic engineering and fermentation process was considered a very prominent technique to synthesize AMPs, due to its potential advantages in low-cost scale-up production. This technique includes the incorporation of the genes of AMPs in suitably chosen expression vectors, including plasmids, cosmids, bacteriophages, and some other novel expression vectors to express these genes in several expression systems like bacteria, yeast, transgenic animals, and plants for the synthesis of AMPs (Wang et al. 2023). However, this technique has also been found inappropriate for the synthesis of AMPs due to the suicidal effects of AMPs on the source microorganisms, resulting in the inhibition of the biosynthesis of the AMPs. Several modern automated methods to synthesize and screen the AMPs and the utilization of heterologous expression systems, such as microbial cells or plant cells, have made it possible for the easy and cost-effective production of AMPs on a large scale (Hoelscher et al. 2022; Nuti et al. 2022; Tanhaieian et al. 2018). The latest study by Nuti et al. has shown the utilization of one of the novel automated techniques, which includes a novel microfluidic system that tends to simultaneously synthesize and screen the AMPs using high-frequency water-in-oil-in-water double emulsion droplets (Nuti et al. 2022). A recent study by Khademi et al. has utilized the technique of genetic engineering to produce novel recombinant antimicrobial peptide by fusing a dermaseptin B1 (DrsB1) peptide with a chitin-binding domain (CBD) from Avr4 protein for Agrobacterium tumefaciens-mediated transformation of tobacco plants. The expression of this novel recombinant peptide DrsB1-CBD in the tobacco plant has shown an increased antifungal activity against fungal pathogens, decreasing the appearance of fungal disease symptoms (Khademi et al. 2020). Genetic engineering techniques for designing and synthesizing novel AMPs also have the potential for development in several upcoming years. Some of the recent advancements in artificial intelligence and machine learning (AI/ML) technologies have revolutionized the process of drug discovery and significantly contributed to the development of rapid and cost-effective designing of drugs, including AMPs (Talat & Khan 2023).
The utilization of AI/ML techniques for the designing of AMPs involves several machine learning and deep learning-trained models for the prediction of potential AMP sequences mentioned in Fig. 3. Some traditional machine learning algorithms, including Support Vector Machines (SVM), random forests (RF), K-nearest neighbors (KNN), and logistic regression, have gained significant importance in the designing of various novel drugs, including AMPs (Ji et al. 2024). The development of Quantitative Structure–Activity Relationship (QSAR) models is a type of novel machine learning approach, which involves the training of these models by analyzing several structural characteristics (descriptors), including cationic nature, helical structure, disulfide bonds, amphiphilicity, etc., of the given train sets of peptides. Further, these trained models are subjected to the test sets of peptides to evaluate their accuracy and then these developed QSAR models are subjected to various online AMP databases, such as APD3, AVPdb, CAMPR3, dbAMP, DBAASP, LAMP2, etc. to predict and identify the potential therapeutic AMP candidates (Bin Hafeez et al. 2021; Cardoso et al. 2020). However, the QSAR models can only identify and classify the available peptides in several databases and cannot generate novel peptides. So, several deep learning approaches based on receptors, ligands, several structural aspects, and random sampling, which involve AI-based algorithms utilizing deep neural networks (DNNs), including convolutional neural networks (CNNs), recurrent neural networks (RNNs), graph convolutional networks (GCNs), and their variants and hybrid models, have been developed to generate novel AMP sequences and this generation of AMP sequences via deep learning is referred as de novo designing of AMPs (Cardoso et al. 2020; Ji et al. 2024). The 2 types of widely utilized AI-based algorithms for the development of deep learning models for the design of AMPs are generative adversarial networks (GANs) and variational autoencoders (VAEs), including their conditional variants (Cao et al. 2023; Das et al. 2020). A recent study by Lin et al. included the AI-based model by training a Wasserstein generative adversarial network with gradient penalty (WGAN-GP) based on existing AMPs to generate novel peptide sequences with antimicrobial efficacy. In this study, 8 GAN-designed peptides were generated by this developed AI-based model and 7 out of these 8 peptides showed potent antimicrobial activity with two of them displaying broad-spectrum antibacterial activity (Lin et al. 2023). Another latest study by Pandi et al. involved the utilization of deep generative variational autoencoders (VAEs), which was composed of an encoder for the peptide sequence input, a latent space for the sampling of the various points, and a decoder for the reconstruction of sequences, to generate novel peptide sequences with potent antimicrobial property. In this research study, the model generated about 500,000 novel peptide sequences, which were then filtered to about 50,000 sequences based on their length and viability. Further, the 500 prior AMP sequences were selected by performing molecular dynamics simulation and synthesizing them for their analysis through the in vitro evaluation tests (Pandi et al. 2023). Several limitations associated with these two AI algorithms are mentioned in one of the recent studies, which include the incomprehensiveness of these trained models, resulting in an unconstrained generation of peptides i.e., these models generate peptide sequences only from the known active AMPs and not from the non-AMP sequences. So, this study included the development of a new model ‘HydrAMP’, which was trained for an analogue generation of peptides i.e., this model can generate peptide sequences from both the active and non-AMP peptide sequences (Szymczak et al. 2023).

Key algorithms in computer-aided design of antimicrobial peptides
In addition to de novo designing of AMPs by deep learning algorithms, several other algorithms, including evolutionary/genetic algorithms, linguistic models, and pattern insertion algorithms, have also significantly contributed to the design and generation of novel AMPs. In evolutionary/genetic algorithms, the models are developed by analyzing the processes of natural evolution and genetic variation of biological samples. These models are trained to generate novel antimicrobial peptide sequences by simulating the process of natural evaluation through genetic mutations in the template AMP sequences (Ji et al. 2024). The development of a linguistic model strategizes the utilization of a set of regular grammar rules, considering each amino acid a “word” and placing it at the appropriate location in the “phrase” (peptide sequence) to make sense. This model analyses the repeated sequences associated with the specific characteristic within the trained datasets of peptide sequences to design novel AMP sequences with improved antimicrobial efficacy. This model can be utilized to identify the particular patterns of sequences present in the AMPs from the various databases available and incorporate them into the template sequences to improve their antimicrobial efficacy. Considering the basis of correlation between the functions of peptides with the patterns of sequences and structural motifs, the pattern insertion algorithms build models to analyze the structural sequences and predict the functional patterns in the set of AMPs, which can be inserted into the template sequences to generate the novel AMP sequences (Cardoso et al. 2020).
Further, these AI/ML approaches can be combined with various modern bioinformatics tools and techniques, including molecular dynamic simulation for their validation (Ji et al. 2024). The utilization of several gene editing tools like CRISPR-Cas technology would also aid in precise changes of the genetic sequences to yield novel AMPs with optimized properties and would increase the rate of designing and synthesizing AMPs. (Wang et al. 2023). In addition, AMPs have also been synthesized by the technique involving the ring-opening polymerization of α-amino acid N-carboxy anhydride (NCA). This technique has been employed particularly, for the synthesis of AMPs incorporated in the star-shaped polymers (Kawano et al. 2020; Vanzolini et al. 2022a).
Mechanisms of Action
Antibacterial Activity
The antimicrobial properties associated with AMPs are mainly due to their cationic and lipophilic nature (Bechinger 2017; Bellotti and Remelli 2022). These unique characteristics of AMPs render their binding to the negatively charged microbial surfaces, which also have high lipid content. The reason behind the specificity of AMPs against microbial surfaces lies in the differences in composition between the cell envelope of the prokaryotes and the eukaryotes (Vanzolini et al. 2022b). The presence of negatively charged constituents, including lipopolysaccharide (LPS) and lipoteichoic acid (LTA), on the cell wall of prokaryotes, renders them the targets for the AMPs with net positive charge. Meanwhile, the eukaryotic cells that possess a neutral charge are spared from the activity of AMPs. The bonding between the AMPs and the microbial cell wall involves the electrostatic interactions between the positively and negatively charged components (Malanovic and Lohner 2016; Zhang and Yang 2022).
Upon attachment to the microbial cell walls, AMPs disrupt the biosynthesis of Peptidoglycan, which is a polymeric component, forming an integral layer of the cell wall (Li et al. 2022). The Peptidoglycan layer plays a vital role in maintaining the integrity of the cell wall and consists of long glycan chains cross-linked together by peptide bonds. The biosynthesis of peptidoglycan includes transglycosylation between the two monosaccharide units, N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG), to form disaccharide, and these disaccharide units constitute the repetitive units that are linked together by transpeptidation (peptide bond linkages) to create the peptidoglycan layer (Walter and Mayer 2019). Lipid II, found on the inner side of the cell membrane, is an important component in the biosynthesis of peptidoglycan, which is responsible for the transportation of cell wall subunits from the inside of the cell membrane to the outside after the transglycosylation reaction for their polymerization by transpeptidation (Le et al. 2017). AMPs are responsible for inhibiting the action of Lipid II, which leads to the disruption in the biosynthesis of peptidoglycans. This disruption in the synthesis of peptidoglycans will disintegrate the cell wall, resulting in cell lysis and expelling out the intracellular contents. Some of the marketed AMPs that target Lipid II include Vancomycin, Oritavancin, Plectasin, Plusbacin-A3(pb-A3), Plectasin, and Tridecaptin A1. Some AMPs like Ramoplanin are responsible for the inhibition of the conversion of Lipid I to Lipid II and thereby disrupt the biosynthesis of peptidoglycan (Li et al. 2022; Münch et al. 2015).
Some AMPs also act by inhibiting the biosynthesis of teichoic acid, which is an acidic polymer present in the cell wall, in addition to peptidoglycan, and is responsible for protecting the bacteria from thermal injury and regulating the movement of ions across the cell wall. Teixobactin is responsible for the inhibition of biosynthesis of the teichoic acid by inhibiting its main precursor Lipid III. Some of the AMPs are also responsible for the destruction of the cell wall by stimulating the release of autolysins, which induce autolysis of the bacterial cell. Pep-5 and Nisin are two AMPs accountable for releasing autolysins by competitively displacing the cell wall-associated amidases (Hussein et al. 2020; Li et al. 2022). AMPs like Thanatin, which is derived from an insect, act on the outer membrane of the cell wall of Gram-negative bacteria, thereby exerting anti-gram-negative bacterial properties. Thanatin is responsible for the formation of aggregates of lipopolysaccharides micelles, thereby disrupting the high lipid-containing outer membrane of the gram-negative cell wall. Along with the aggregate formation in the outer membrane, Thanatin also displaces the Ca2+ ions that are bound to LPS molecules and further contributes to the destruction of the outer membrane (Dash and Bhattacharjya 2021).
After attaching and disrupting the cell wall, the AMPs get into the cell membrane of pathogenic microorganisms by physicochemical interactions, which depend on two main factors; peptide-lipid ratio and conformational changes in the structure of the peptides (Li et al. 2022; Zhang et al. 2021a, b). AMPs are arranged parallelly with the surface of the cell membranes if the peptide-lipid ratio is low, whereas, in the case of a high peptide-lipid ratio, AMPs can directly penetrate the cell membrane. Structural conformations of the peptides also affect the binding affinity of the AMPs. The AMPs having α-helical structures, like LL-37, cecropin, and magainin, bind more effectively than other conformations, as they can bind the cell membranes even at low peptide-lipid ratio (Maleki Dizaj et al. 2022a).
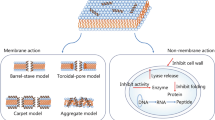
The mechanism of the interactions and disruption of the cell membrane by the AMPs can be explained by the 3 main models; barrel-stave, carpet-like, and toroidal pore models as shown in the Fig. 4. (Bellotti and Remelli 2022). The barrel-stave model explains the formation of aggregates due to the interactions between the hydrophobic regions of the peptides and the lipidic domains of the cell membrane, which leads to the perturbation of the membrane parallelly, resulting in the formation of transmembrane pores, which eventually damages the membrane and leads to the leaching of the cellular contents and thereby causing cell lysis. The AMPs acting through the barrel-stave model are Alamethicin and Ceratotoxins (Li et al. 2022). In the carpet-like model, the AMPs interact electrostatically with the negatively charged components cell membrane and start to accumulate in the cell membrane. As the concentration of the AMPs in the cell membrane reaches the threshold value, they disrupt the cell membrane by rupturing it. AMPs like Magainin and Citropin are responsible for the cell membrane disruption using a carpet-like model. The toroidal pore or wormhole model involves the perpendicular accumulation of the AMPs in the cell wall, resulting in the narrowing of the lipid portion and the formation of the nanometric size circular pore within the cell membrane, which eventually leads to the disruption in the cell membrane. Melittin, Actinoporins, and Lacticin Q are the AMPs acting via the toroidal-pore model. Some AMPs, like Protegrins, act via multiple models to disrupt the cell membrane by barrel-stave as well as the toroidal-pore model and Magainin disrupts by the carpet-like as well as the toroidal-pore model (Bellotti and Remelli 2022; Li et al. 2022). The AMPs also interact and disrupt the cell membrane by numerous mechanisms that have been proposed and these include receptor-mediated disruption mechanism by activating the stress-response pathways (Nisin) (Shin et al. 2016), ROS (reactive oxygen species) damaging mechanism that increases the oxidative stress, thereby inducing cell apoptosis (Psacotheasin) (Zhang and Yang 2022). etc.

Mechanisms of action and immunomodulatory effects of antibacterial peptides (amps)
Cell-penetrating AMPs can enter within the cell by crossing the cell envelope to exert the antimicrobial effect on the intracellular targets, including the nucleus, cell organelles, several enzymes, and other proteins within the cell (Mukhopadhyay et al. 2020; Zhang and Yang 2022). The transportation mechanism of these intracellular-acting AMPs across the cell envelope has not been comprehensively understood. Some studies have stated several mechanisms through which these AMPs can enter the cell, and these include direct penetration by the formation of transient toroidal gaps within the cell envelope or by direct translocation through the defects within the cell envelope, endocytosis, or using other cellular delivery systems like receptor-mediated transport pathways (Li et al. 2022; Luo & Song 2021). Intracellularly, AMPs act on different sites through various mechanisms, including binding and modulating of nucleic acids, inhibition of intracellular enzymes and other vital proteins, interruption in the arrangement and assembly of protein structures, and inhibition of the activity of various cell organelles (Vanzolini et al. 2022b). Actions of the AMPs by binding the nucleic acids lead to the modulation or destruction of DNA and RNA conformations, which may inhibit their synthesis. These changes would also result in the inhibition of protein synthesis or may result in the synthesis of defective proteins by altering the transcription or the translation process (Li et al. 2022). Buforin-II, Indolicidin, PR-39, Human neutrophil peptide (HNP)-1, porcine β-defensin 2 (pBD2), and Rondonin are some of the AMPs acting by binding to nucleic acids (Li et al. 2022).
Inhibition of enzymes and other vital protein substrates by AMPs may lead to numerous alterations in the normal physiology of the cell, including inhibition in the DNA and protein synthesis, inhibition of several important metabolic pathways, interruption in the cell division process, destruction of nucleic acid damage repair mechanism, etc. (Li et al. 2022). Some of the AMPs acting via inhibiting the enzymes and other vital protein substrates are Microcin (Mcc) B17 and J25 (inhibit DNA gyrase and RNA polymerase, respectively) (Collin & Maxwell 2019; Galván et al. 2019), Indocilidin (targets DNA topoisomerase I, thereby inhibiting DNA relaxation) (Rokitskaya et al. 2011), Temporin L (inhibits FtsZ protein, resulting into the interruption of cell division process and thereby blocking cell cycle) (Di Somma et al. 2021), Histatins (histidine-rich peptide) (Li et al. 2022), Tritrpticin (tryptophan-rich peptide) (Shagaghi et al. 2016), Apidaecin (inhibits the action of polypeptide release factors on 70S ribosomes) (Graf & Wilson 2019). Some AMPs are responsible for inhibiting the activity of various cellular organelles such as ribosomes, mitochondria, vacuoles, etc. Proline-rich AMPs (PrAMPs) like Bac7, Pyrrhocoricin, Drosocin, and apidaecin bind to the ribosome and interrupt the early stages of the translation process, thereby inhibiting the protein synthesis (Gagnon et al. 2016; Graf & Wilson 2019). AMPs altering mitochondrial activities, like depolarization of the mitochondrial membrane, leads to significant changes in cell physiology (Zhang & Yang 2022). PrAMPs have also been found to interrupt the arrangement (folding) and assembly of the protein structures by inhibiting the DnaK protein that is responsible for the proper arrangement of the protein structure (Knappe et al. 2016; Vanzolini et al. 2022b).
Antifungal Activity
AMPs showing antifungal activity, bind to the cell wall of fungi and inhibit the biosynthesis of fungal cell wall components like glucan, chitin, and mannan. Glucan inhibitors like Echinocandin, Caspofungin, and Micafungin can noncompetitively inhibit glucan synthesis (Li et al. 2021). A nucleoside containing dipeptide, Nikkomycin Z, which is synthesized by Streptomyces tendae, acts by inhibiting the biosynthesis of the chitin in the fungal cell wall and thereby disrupting it (Kovács et al. 2019). Mannan and its conjugates are the important constituents in the synthesis of biofilm, which is a fungal virulence factor responsible for the adaptations in the fungal cells to evade the host’s immune system. Biofilms also render the fungal cells multidrug resistant by protecting against various antifungal drugs. So, the AMPs that act by binding to mannan, like Pradimicins and Benanomicins, form complexes with the calcium and lead to the inhibition of the synthesis of biofilm, thereby disrupting the fungal structural integrity and killing the fungal cell. In addition to inhibiting biofilm synthesis, some antifungal AMPs also act by eradicating the mature biofilms(De Cesare et al. 2020; Vanzolini et al. 2022b). Some AMPs like AMP-17 also disrupt the fungal cell wall by acting on ergosterol synthesis-related genes and inducing their downregulated expression, which affects the biosynthesis of the ergosterol and results in the disruption of the cell wall (Ma et al. 2020).
Due to its eukaryotic nature, fungal cell membranes have high similarities with the mammalian cell membrane, making it difficult to target the fungal cell membrane. Few AMPs with synthetic modifications have been found to improve their safety and efficacy by targeted delivery (Vanzolini et al. 2022b). Though the mechanism of action of the antifungal has not been studied comprehensively, these AMPs have been found to act by the same antimicrobial mechanism models (De Cesare et al. 2020). Some of the AMPs also interact with several membrane components like glucosylceramides and β-glucans and with some of the enzymes responsible for synthesizing other vital cell membrane components (Vanzolini et al. 2022b).
The intracellular targets of AMPs involved in fungi include nucleic acids and cell organelles. KW4 is a synthetic peptide that has been found to inhibit cellular functions by binding and destroying the DNA and RNA (Ramamourthy et al. 2020). Some AMPs acting intracellularly can also interact with vital proteins and cell organelles (particularly mitochondria and vacuole), resulting in the change of fungal cell morphology. The fungal cell envelope also consists of several translocators like siderophore iron transporter 1 that can transport the AMPs within the cell (Nakamura et al. 2019; Vanzolini et al. 2022b).
Antiviral Activity
Viruses are extremely small microorganisms that cannot replicate on their own, but they can utilize the host cell’s enzymes and other proteins for their proliferation within the host cell, thereby resulting in infectious disease to the host. The viral replication cycle in the host consists of several steps, including attachment, penetration, uncoating, replication, assembly, maturation, and release (Ryu 2017). The inhibition of any step in the viral replication cycle results in the inhibition of the growth of the virus within the host (Fig. 5). Peptides acting against the virus are referred to as “Antiviral peptides (AVPs),” and AVPs act via different mechanisms to kill and inhibit the virus growth within the host (Vanzolini et al. 2022b; Vilas Boas et al. 2019).

Mechanism of action of antiviral peptides and lifecycle of a virion
Some AVPs are found to directly act on the virus particles by damaging and killing them before entering the host cell (an extracellular mechanism). The mechanism behind the direct action on viral particles involves the interactions of AVPs with the viral envelope to damage several surface particles, thereby resulting in its disruption and instability. This action also inhibits the interaction of the virus with the host’s cell membrane receptor and prevents the virus from causing the infection (Li et al. 2022; Maiti 2020). Enfuvirtide is an AVP that acts by inhibiting the glycoprotein 41 on the HIV-1 (Human Immunodeficiency Virus 1) and prevents the attachment of viral particles with the host cell membrane, thereby preventing the spread of HIV infection (Eggink et al. 2019). Mucroporin-M1, DN59, LL-37, and Gloverin are some of the other AVPs responsible for damaging the viral envelope and leading to the inhibition of various viral infections (Diamond et al. 2021). The extracellular action of AVPs also involves inhibiting the virus’s uncoating process upon attachment to the host cell. P9 and P9R, the derivatives of β-defensin 4, are found to block the process of virus uncoating by the inhibition of late endosomal acidification, which is important for the viral and the host endosomal membrane fusion (Li et al. 2022; Zhao et al. 2020).
Some AVPs act through intracellular mechanism (after entering within the host cell) by inhibiting the various stages of the intracellular viral life cycle, including viral gene expression and replication, virus assembly, virus release from the host cell or interference with the intracellular signaling pathways required for the replication of the virus within the host cell (Li et al. 2022). AVPs like HNP2 and HD5 of the α-defensins class have been found to bind with the DNA and block the gene expression in HSV-2 (Herpes simplex virus-2) (Xu & Lu 2020). LL-37 can act through both the mechanisms, extracellular as well as intracellular, in a dose-dependent manner. Intracellularly, LL-37 inhibits reverse transcription by inhibiting reverse transcriptase activity, thereby inhibiting the viral gene expression and replication (Li et al. 2022; Tripathi et al. 2015). Infections by many viruses need the activation of intracellular signaling pathways involving PKC (Protein kinase) for the regulation of processes involved in various steps of the viral replication cycle, including viral integration, aggregation, and release. Inhibition of these PKC-activating intracellular pathways leads to the interruption of the viral replication cycle. HNP1 acts by inhibiting the activation of PKC in primary CD4 + T cells to block the HIV-1 infection (Solanki et al. 2021). Some AVPs also interrupt other signaling pathways to inhibit the viral infection. AVPs like GF-17, BMAP-18, and Smp-76 (Scorpion Venom Peptide) have been found to act on the interferon signaling pathways to inhibit various viral infections (He et al. 2018; Ji et al. 2018).
Immunomodulatory Actions of AMPs
In addition to antibacterial, antifungal, and antiviral activities, AMPs can also modulate immune responses within the host. Immunomodulatory AMPs, like AMPs of the human β-defensin (hBD) class, display chemokine-like activities and can chemoattract leukocytes via chemokine receptors to elicit the immune responses at the infected site (Zhang & Yang 2022). AMPs of the hBD class also act by modulating the pro-inflammatory reactions through the inhibition of LPS-induced pro-inflammatory cytokines like IL-6, IL-1β, and TNF-α and inhibition of Toll-like receptors on activated macrophages that are responsible for mediating the cytokine-induced pro-inflammatory responses (Zhang & Yang 2022). LL-37 is accountable for modulating the innate immune system and T-cell differentiation. It also acts by up-regulating the expression of interferon and cytokines (attracting antigen-presenting cells), thereby modulating the antiviral immune system to inhibit the viral activities within the host cell (Pahar et al. 2020).
Strengths and Limitations
AMPs have been found to have numerous advantages over the older antibiotics. Some advantages of using AMPs over older antibiotics are mentioned here (Table 1). AMPs have fast-killing mechanisms and higher potency in inhibiting pathogenic microorganisms than older antibiotics. The utilization of AMPs is more advantageous, as they have broad spectrum anti-microbial properties and can act against multidrug-resistant species, which have developed resistance against the older antibiotics, especially the ESKAPE group, which refers to a group of six MDR pathogens, including Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species (Moretta et al. 2021b; Mukhopadhyay et al. 2020). AMPs are also found to develop slowly, or in some cases avoid, microbial resistance against these pathogenic microorganisms compared to older antibiotics due to their unique mechanisms of action to kill the organisms like an attack on the lipid components of the cell envelope, disruption, and inhibition of synthesis of biofilm, stimulating immunomodulatory responses against the microorganisms within the host, etc., which renders the microorganisms inefficient in building the resistance against the AMPs; therefore, AMPs have significant application prospects compared to them (Rima et al. 2021; Wang et al. 2016). Along with the anti-microbial activities, AMPs have also been found to act on diverse targets within the host and contribute to other host-beneficial activities like immunomodulatory activity, wound-healing properties, anti-cancer activity, tissue repair, inflammation, and in some other disease states (Li et al. 2022). Another advantage associated with using AMPs as therapeutic agents is their safety, as their structures closely resemble the endogenous molecules, and their degradation into natural amino acids results in a very low adverse drug response and minimized side effects. The short half-life of AMPs, resulting from their rapid metabolism, rare accumulation within the body tissues, and achievement of equal or higher therapeutic effects at low doses, also contributes to low toxicity. Several AMPs may also contribute to replacement therapies to treat the deficiency or low levels of various endogenous peptides within the body. AMPs have also been found to synergistically enhance anti-microbial activity and reduce drug resistance of the older antibiotics when administered in combination with each other (Lau & Dunn 2018; Mahlapuu et al. 2020).
Though AMPs display potential advantages and have been found to have numerous applications in the treatment of various infectious diseases, but still, only a handful of AMPs, out of several thousand isolated over a decade, have entered the preclinical trials and have further headed toward the clinical studies. The reason behind this issue is that along with the potential advantages of AMPs, they are also associated with several limitations responsible for limiting the clinical applications of AMPs (Bellotti & Remelli 2022). The first limitation that restricts the clinical application of AMPs is their inadequate oral bioavailability. Inadequate oral bioavailability results from factors such as enzyme degradation, lower stability at gastric pH levels, hydrophilicity, high molecular weight, and high salt concentration. All proteins and peptides ingested are susceptible to digestive proteases that are found in the stomach and small intestine. Peptides are also found to be stable at a very limited pH range that is close to their isoelectric pH value. Some AMPs are also found to be inactivated in the gastric pH due to pH-induced unfolding. Moreover, the activity of proteolytic digestive enzymes is also pH-dependent. Pepsin has the strongest potency to degrade proteins and peptides at a gastric pH range of 2–3 but is inactive at pH above 5, whereas trypsin and chymotrypsin are more efficient at an intestinal pH range of 6.5–7.5. The absorption of some AMPs along the gastrointestinal tract is also restricted due to their high molecular weight and hydrophilic nature, resulting in difficulty in passing through the cell membrane of gastrointestinal endothelium. Higher physiological salt concentration may also lead to an inactivation of several AMPs, rendering them ineffective (Zhu et al. 2021). The systemic route of administration is also considered inappropriate for numerous AMPs because of their short half-life within the bloodstream. The short half-life of AMPs results from their inactivation due to serum binding and their susceptibility to proteolytic actions of different circulatory proteases, resulting in their degradation and rapid elimination via renal and hepatic clearance. Additionally, injection-based routes of administration have also been reported to develop allergic reactions and certain infections at the site of administration. Therefore, local administration is considered the favorable route of administration for numerous AMPs, but still, this route of administration poses the threat of degradation by tissue proteolytic enzymes (Greco et al. 2020; Mahlapuu et al. 2020). Despite the higher safety profile as compared to conventional antibiotics, many AMPs are found to have a lower specificity toward their targets and act on the eukaryotic cell membranes to induce cytotoxic responses by degrading them, particularly the hemolytic activity against the erythrocytes and the damaging reactions to the kidneys and the central nervous system (Greco et al. 2020; Yang et al. 2023).
Another limitation associated with AMPs is the development of resistance. However, in comparison to conventional antibiotics, the emergence of antimicrobial resistance to AMPs is less common. However, the slow growth of antimicrobial resistance against AMPs still contributes to a significant problem and will be in the future. The development of antimicrobial resistance has been mostly observed due to the frequent exposure to AMPs and the consequent evolutionary progress of microorganisms by developing various antimicrobial-resistant mechanisms to prevail over the antimicrobial mechanisms of AMPs (Joo et al. 2016; Vanzolini et al. 2022a). Some of the important resistance mechanisms developed by microorganisms to combat the antimicrobial activity of AMPs are alterations in the cell envelope, biofilm formation, the release of proteolytic enzymes, efflux pumps, and several membrane transport systems, sequestration and inactivation of AMPs, and expression/repression of a gene induced by AMPs (Li et al. 2023).
The cell envelope has been altered through various mechanisms to induce resistance against AMPs, including the cell wall, cell membrane, and lipopolysaccharide layer modifications in multiple microorganisms. The resistance due to the changes in the cell wall has resulted from various modifying processes such as the D-alanylation of the wall lipoteichoic acids in several Gram-positive species, including Bacilli spp., Staphylococci spp., Enterococci spp., and Streptococci spp.; the glycosylation (B. cereus and L. monocytogenes) or esterification (staphylococci spp.) of the cell wall teichoic acids; N-deacetylation of the peptidoglycan monomeric constituents like N-acetylglucosamine and N-acetylmuramic acid, as observed in Pneumococci spp. and B. subtilis; and capsule formation around the cell wall, observed in Pneumococci spp. and several Gram-negative species like Klebsiella pneumoniae, Neisseria meningitides, and P. aeruginosa. The modifications in the structure of the cell membrane are the consequences of several membrane-altering mechanisms like the changes in the lipid composition of the membrane by synthesizing lysinylated (Staphylococci spp., Bacilli spp., and enterococci spp.) or palmitoylated (S. typhimurium) phosphatidylglycerols components, and carotenoids pigments called staphyloxanthin (Staphylococci spp.) within the membrane, which leads to the increase in the hydrophobicity and the charge group on the cell membrane, resulting in the repulsion of AMPs due to their cationic and hydrophilic nature and inhibit their binding to the cell membrane (Assoni et al. 2020; Band & Weiss 2014). The outer lipopolysaccharide (LPS) layer in several Gram-negative species has also been found to alter its structure by attenuating the negative charge on the lipid A component through the addition of positively charged molecules, such as cationic sugar moieties like aminoarabinose in P. aeruginosa, Salmonella typhimurium, glucosamine in Bordetella pertussis (Shah et al. 2014); phosphoethanolamine residues in Neisseria gonorrhoeae, Salmonella typhimurium, and A. baumannii (Band & Weiss 2014). Some Gram-negative species like Porphyromonas gingivalis, Bacteroides fragilis, Helicobacter pylori, and F. tularensis have also been found to attenuate the negative charge by removing the negatively charged residues, especially the anionic phosphate group, from the lipid A component of the outer LPS layer. Along with the modification in the lipid A component, the O-antigen and the core sugar residues of the outer LPS layer have also been altered and contributed to the AMP resistance (Band & Weiss 2014).
Some microbes have also been found to develop resistance against antimicrobial compounds by synthesizing biofilms, which are highly organized structures responsible for protecting the microorganism in stressful conditions. Within the biofilm, the microorganisms secrete the extracellular jelly-like substance over their surface to cover the gap between the inner biofilm layer and the surface of the organisms and these mechanisms togetherly protect the microorganisms within the biofilm (Jolivet-Gougeon & Bonnaure-Mallet 2014). However, most of the AMPs have been found to act potently against biofilm-synthesizing microorganisms by inhibiting biofilm synthesis or by directly disrupting the matured biofilm. But still, some of the AMPs have been found ineffective against biofilm-synthesizing organisms (De La Fuente-Núñez et al. 2015). Another mechanism by which the pathogens combat AMPs is by releasing proteolytic enzymes to degrade the AMPs. Some species of Staphylococci, Enterococci and Streptococci genera, along with some Gram-negative species like P. aeruginosa, Proteus mirabilis, S. typhimurium, Shigella flexneri, and Yersinia pestis have found to release various proteolytic enzymes to combat against AMPs, while some microorganisms like P. aeruginosa have also been found to take advantage of the host proteolytic enzymes to degrade the AMPs that acts against them (Assoni et al. 2020; Band & Weiss 2014). Several trans-membrane systems and efflux pumps have also been found to contribute to the development of resistance in numerous microbial species against AMPs. Efflux pumps are the complexes present on the cell membrane that tend to remove the toxic material, along with antimicrobial agents, from the cell. AMPs that act intracellularly and cross the surface barriers can be thrown out of the cell by these complexes and other trans-membrane systems like ABC transporters (Dintner et al. 2011). Some of the species from Staphylococci, group B Streptococci, and Pneumococci genera, along with K. pneumoniae, and P. aeruginosa tend to sequester and inactivate the AMPs by releasing some negatively charged moieties that tend to interact with the cationic AMPs, thereby preventing their attack on the microbial cells (Assoni et al. 2020). Similarly, Gram-negative species like E. coli and V. cholerae are responsible for developing resistance against AMPs by releasing the negatively charged outer membrane vesicles (OMV), which are the outer membrane carriers that carry the intracellular toxic material out of the cell. OMV release also can bind and sequester the AMPs, thereby significantly increasing the resistance levels of these microbes. AMPs acting intracellularly, especially on nuclear material, have also developed resistance due to the induction of gene expression or repression mechanism alteration within various microorganisms (Band & Weiss 2014).
Possible Solutions to Overcome the Limitations of AMPs
The antimicrobial activity of AMPs can be optimized by maintaining the proper levels of their certain characteristics, including hydrophobicity, cationic nature, and structural conformations. Optimization of these characteristics renders AMPs more efficient, stable, and specific (Haney et al. 2017). Apart from optimizing these characteristics of AMPs, the physiological conditions of the GIT tract have to be modulated to enhance the bioavailability of AMPs (Zhu et al. 2021). Various modern technological developments in the medical and pharmaceutical fields can significantly contribute to maintaining the balance between these characteristics, thereby overcoming the limitations associated with AMPs. To date, several modern strategies as per Fig. 6, including modifications to the structure of AMPs, prodrug approach, utilization of novel delivery systems and nanotechnology, pH modulation of GIT, coadministration with protease inhibitors, etc., have been utilized that helped to maintain the proper balance of the attributes and overcome the limitations of AMPs (Mukhopadhyay et al. 2020; Zhu et al. 2021).

Strategies to Overcome the Limitations of Antimicrobial Peptides (AMPs)
Structural Modifications
The modifications in the structure of AMPs have been done by utilizing several strategies that involve alterations in amino acid sequences, synthesis of peptidomimetics, end-terminal modifications, cyclization, multimerization, and hybridization (Zhang et al. 2021a, b).
Sequential Modifications of Amino Acids
Modifying amino acids from the natural sequences of AMPs is the most frequently used technique to overcome their limitations. The amino acid sequences can be modified by either adding, deleting, or substituting the amino acids in the natural sequence of AMPs. These modifications can result in the optimization of certain attributes of natural AMPs like an increase of cationic character, optimizing hydrophobicity, and changes in the structural conformations (α-helical, β sheet, and cyclized), thereby reducing cytotoxicity, lowering proteolytic degradability, increasing the efficacy and selectivity against their target (Bellotti & Remelli 2022; Zhang & Yang 2022). KR-12, an analog of LL-37, which was developed by deleting the Phe residue at the 17th position in the LL-37 sequence, decreased cytotoxic responses against the host cells without lowering the antimicrobial efficacy (Ting et al. 2020). Eliminating more susceptible amino acids to protease degradation from the peptide sequences has also resulted in forming AMPs with high tolerability against proteolytic degradation (Bellotti & Remelli 2022). The substitution involves the residual substitution of one or more amino acid residues with different amino acid residues in the natural amino acids sequence of AMPs. A research study on Anal 3 has shown the formation of Pro-hinge in Anal 3 by substitution of Glu by Pro at 9th position in the natural amino acid sequence of Anal 3, which resulted in an increase in peptide selectivity against the microorganisms, thereby lowering the cytotoxic responses within the host (Ting et al. 2020). The substitution with the hydrophobic residues has also been found to increase the hydrophobicity of the less hydrophobic AMPs, thereby enhancing their membrane penetrating power (Chegini et al. 2019). The isomeric modification of AMPs, obtained by adding or substituting the natural amino acids with their enantiomers, L- to D-substitution, has been shown to considerably reduce the degradation of AMPs by the proteolytic enzymes, as the tolerability of D-amino acids to these enzymes is greater as compared to L-isomers, thereby enhancing their activity and selectivity towards the targets (Zhang et al. 2021a, b). The isomeric modification of Daptomycin led to its approval by the United States Food and Drug Administration (USFDA) against several gram-positive microorganisms involved in various dermal and systemic infections (Qvit et al. 2017). However, this is a sequence-dependent strategy, suggesting that this effect makes specific conformational changes in the structure of the peptides, depending on their sequences, and does not apply to all the peptide sequences (Lu et al. 2020).
Peptidomimetics Approach
In addition to the substitution with the proteogenic amino acids (20 amino acids), different non-proteogenic amino acids like ornithine, 2,4-diamino-butyric acid (DAB), and 2,3-diamino propionic acid (DAP), L-4,5,6,7-tetrahydro-1H-imidazo [4,5-c] pyridine 6-carboxylic acid (Spi), L-1,2,3,4-tetra-hydro-isoquinoline-3-carboxylic acid (Tic), 4-amino butanoic acid, 2,3-diamino propionic acid (DAP), palmitic acid, etc. have also been utilized, resulting in the synthesis of peptidomimetics/pseudo peptides with enhanced cationic character and nonproteolytic amino acid residues, there by enhancing antimicrobial efficacy, stability, and selectivity, compared to the natural AMPs (Arias et al. 2018; Ting et al. 2020). The substitution with the rigid derivatives of proteogenic amino acids, including β-didehydrophenylalanine, 2-naphthyl-L-alanine (an aromatic residue), S-tert-butylthio-L-cysteine, etc., has also been employed in the substitution of natural amino acids in the AMPs (Oliva et al. 2018a). The modifications in the peptide backbone in the structure of natural AMPs have also resulted in the synthesis of peptidomimetics(Wątły et al. 2021). Several strategies that have been employed in the modification of the peptide backbone in the structure of natural AMPs include the alkylation of the nitrogen at the N-terminal, reduction of the carbonyl group into the methylene group, the substitution of the –NH– group with the other electronegative groups like oxygen (–CO–O–), sulfur (–CO–S–), or methylene group (–CO–CH2–), and substitution of the α-CH group with the nitrogen atom, thereby inducing the β-sheet in the structure of the peptide(Wątły et al. 2021).
End-Terminal Modifications
End-terminal modifications (N-and C-terminal modifications) are naturally observed in various eukaryotic and prokaryotic species to modify certain proteins and peptides and enhance their activity and stability (Oliva et al. 2018b). Several host defensive peptides (HDP) have also been observed to undergo natural end-terminal modifications in various species, resulting in their enhanced activity and stability (Dahiya et al. 2018). Aurin, Melittin, Modelin-5, etc., are observed to undergo C-terminal amidation naturally, which leads to greater α-helical stability at the peptide-membrane interfaces, resulting in the greater binding and destruction of the membrane, thereby enhancing the antimicrobial efficacy of these peptides (Mura et al. 2016). In contrast to C-terminal modifications, N-terminal modifications like N-acetylation are also frequently observed in various species naturally (Ree et al. 2018). MreB, the HDP derived from E. coli, has effectively killed the microorganisms in higher salt concentrations, inferencing the significant reduction in the salt sensitivity of this peptide(Saikia et al. 2017). Along with these end-terminal modifications, some other end-terminal modifications like N-methylation (Dahiya et al. 2018), N-pyroglutamate(Nath et al. 2023), N-terminal myristoylation by conjugating myristic acid (Liu et al. 2020), conjugation of cholesterol at N-terminal (Chen et al. 2020), and conjugation of 6-aminocaproic acid (Purwin et al. 2017) at either both of the terminals have been studied. Unfortunately, end-terminal modifications have also resulted in a decrease in the antimicrobial efficacy in some of the AMPs because of the overall reduction of the positive charge in their structures, inferencing that this modification strategy is sequence-selective and not all the peptides can benefit by utilizing it (Soleymani-Goloujeh et al. 2018).
Cyclization
Another important modification strategy to overcome the limitations of AMPs is cyclization, which involves the ligation of the amino and the carboxy terminals with each other, thereby concealing these ends, to produce a cyclic peptide with increased stability and specificity (Conibear et al. 2016). Cyclization is a natural phenomenon found in many species to enhance the strength and specificity of peptides, including HDPs (Zhang et al. 2018). Cyclization has also improved antimicrobial efficacy by forming the β-sheet structure of AMPs (Gunasekera et al. 2020). So far, cyclization has also been utilized in vitro to increase the stability and specificity of multiple AMPs, including vancomycin, daptomycin, and colistin/polymyxin (Ting et al. 2020). Apart from the end-to-end ligation of peptides, cyclization can also be achieved by the ligation of two sidechains with each other or the sidechains with the end-terminals or by intramolecular cross-linking between cysteine residues due to the formation of disulfide bonds within these residues (Scudiero et al. 2015). A recent study on the development of novel cyclic AMP ZY4, which was developed by the introduction of the disulphide bond in the natural HDP cathelicidin-BF, has been found to have higher proteolytic stability and antimicrobial efficacy against MDR P. aeruginosa and A. baumannii, as compared to the parent peptide (Mwangi et al. 2019). Sometimes, the ligation of sidechains may result in the formation of the α-helical structure of the peptides, which may also contribute to higher specificity and antimicrobial efficacy of the AMPs. This technique of forming the α-helical structure by ligating sidechains is known as stapling (Moiola et al. 2019). The latest analytical report on designing and developing novel derivatives of magainin-2 by stapling between the sidechain and the N-terminal has been shown to enhance the antimicrobial efficacy against various pathogenic species with greatly reduced hemolytic activity compared to magainin-2 (Hirano et al. 2021).
Multimerization and Hybridization
Natural multimerization, i.e., the combination of two or more peptides, has been found to exert a synergistic effect against various microorganisms to protect the host. This strategy was then greatly utilized in vitro to combine several AMPs, which resulted in the synergistic effect to produce a stronger antimicrobial effect with broad-spectral activity against various microorganisms (Ting et al. 2020). Later on, the utilization of this strategy was gradually reduced due to certain disadvantages associated with this technique, like an increase in host cytotoxicity, high synthesis cost, and increased peptide length, rendering it difficult for the peptide to cross the cell membrane (Grassi et al. 2017). However, recent studies have again shown the utilization of this technique, involving the combination of the AMPs with other pharmacokinetic-and pharmacodynamic-enhancing peptides such as cell-penetrating peptides (CPPs) (Kim et al. 2021). CPPs are members of the class of membrane-active peptides that can translocate within the cells without significantly altering the cell membrane (Bahnsen et al. 2015; Zhang et al. 2021a, b). One of the recent studies on the development of a novel peptide conjugate from the combination of the CPP named ‘R9’ with magainin and M15 has been found to show the increased 2–4 folds greater antimicrobial efficacy against Gram-positive species and 4–16 folds against Gram-negative species, as compared to the parent peptides (Lee et al. 2019). Another study by Drexelius et al. hypothesized that CPPs should be directed towards antimicrobial activity based on the rationale of several similar physicochemical characteristics between CPPs and AMPs. This study has shown the optimization of the structure of CPPs C18 by structure-based sequence analysis to identify the active residues within a CPP sequence and to design and develop highly efficient novel AMPs with greater membrane-penetrating properties (Drexelius et al. 2021). Later, the development of several modern techniques, including the development of peptide sequencing methods, computational databases, and various in silico analytical techniques that had contributed to a comprehensive understanding of the peptide sequences, led to the development of another novel sequence-based strategy, named hybridization, which had the potential to overcome the disadvantages associated with the multimerization technique (Almaaytah et al. 2018; Wade et al. 2019). In this technique, the active amino acid residues from multiple (2–3) AMPs with different modes of action are merged to produce a single sequence (Wang et al. 2019). After developing this technique, many novel hybrid peptides have been synthesized. A recent study on the design and development of a novel hybrid AMP named ‘BMR-1’, which was synthesized by using the active amino acid residues from the Frog Esculentin-1a and Monkey Rhesus cathelicidin (RL-37), has shown to have optimized physicochemical parameters with enhanced surface charge and this resulted in an increased antimicrobial efficacy of the newly synthesized BMR-1 against various multidrug-resistant Gram-negative species (Tall et al. 2020). Another research on the design and development of novel trihybrid AMPs named CaLL, CaMA, LLaMA, and MALL, which were synthesized from cecropin A, LL-37, and magainin II, has also shown greater antimicrobial efficacy as compared to their parent AMPs (Ting et al. 2020). Utilizing this rational-based hybridization strategy in designing and developing novel peptide sequences has also been shown to reduce other limitations associated with AMPs, like cellular permeability, instability in higher salt concentration and serum, and cytotoxicity. Hence, this strategy has been proven highly potent for synthesizing novel hybrid AMPs with greater therapeutic activity and availability compared to parent peptides (David et al. 2018).
Prodrug Approach
The utilization of the prodrug approach has also been beneficial in overcoming the limitations associated with AMPs, which include the chemical derivatization or conjugation of peptides with different biodegradable chemical agents (Renukuntla et al. 2013). Mainly, the prodrug approach involved the conjugation of AMPs with several formulation polymers, including polyethylene glycol (PEG), poly lactic-co-glycolic acid (PLGA), poly-L lysine (PLL), and hyperbranched polyglycerol (HPG) (Maleki Dizaj et al. 2022b; Zhang et al. 2019). The method of conjugation with PEG is known as PEGylation, and this approach has been widely studied and extensively utilized to overcome the limitations of AMPs. This technique involves the covalent binding of a short chain of PEG polymer to the peptide sequence of AMPs, particularly at the C-terminal (Mahlapuu et al. 2020). A study on the PEGylation of M33 at the C-terminal had shown to decrease proteolytic degradation in the presence of P. aeruginosa elastase by increasing resistance against it (Falciani et al. 2014). Polymeric conjugations have also been utilized for sustained release and site-specific drug delivery of AMPs, ensuring their precise loading on the targeted microorganism. Some polymers have also been found to direct the AMPs across cellular membranes to act on the intracellular targets (Cui et al. 2021). A recent study on the conjugation of PLGA with esculentin-1a-derived AMP was shown to prolong the therapeutic efficacy of esculentin-1a-derived AMP against Pseudomonas aeruginosa lung infection (Casciaro et al. 2019). Apart from conjugating with polymers, the prodrug approach also includes reversible lipidization and conjugation with organometallic agents like ferrocene. Lipidization enhances the membrane permeability of AMPs by increasing their hydrophobicity, but it has been associated with the limitation of decreasing the efficacy of some AMPs. So, reversible lipidization has been employed to overcome this limitation (Zhu et al. 2021). Conjugation of AMPs with organometallic agents results in the formation of organometallic AMPs (OM-AMPs), which have shown significantly increased antimicrobial efficacy against various pathogenic species (Albada & Metzler-Nolte 2017).
Novel Drug Delivery Systems
The bioavailability of AMPs can also be improved by utilizing novel drug delivery systems. Various studies have successfully found that certain limitations associated with AMPs can be overcome by using novel delivery approaches, including nanotechnology (Carratalá et al. 2020). Nano-delivery systems have been found to deliver AMPs via two mechanisms, which include passive delivery (encapsulation within nanocarriers) and active delivery (self-assembling on the surface of nanocarriers) (Schnaider et al. 2020). Utilization of several nanocarriers such as liposomes, micelles, metallic nanoparticles, polymeric nanoparticles and nanofibers, mesoporous nanoparticles, carbon nanotubes, hydrogels, and dendrimers has been found beneficial in enhancing the antimicrobial efficacy, stability and specificity of various AMPs (Maleki Dizaj et al. 2022b; Nordström & Malmsten 2017).
Liposomes
Liposomes are round vesicular bodies consisting of a bilayer of lipid molecules, which can entrap hydrophobic as well as hydrophilic drugs in it, and they have been best suitable for the delivery of hydrophilic and amphiphilic AMPs. Liposomes have been found to increase the half-life and reduce the cytotoxicity of various AMPs by enhancing their proteolytic stability and targeted delivery (Allahou et al. 2021). However, this delivery system has also been associated with several disadvantages, such as some AMPs being found to interact with liposomal surfaces to cause their disruption, liposomes interacting with platelets and leading to thrombocytopenia, and difficulty in large-scale production of liposomes, which limits their clinical applications (Jash et al. 2021).
Micelles
Like liposomes, micelles are also round vesicular bodies but are small in size and are composed of surfactants or other amphiphilic molecules. These constituent molecules are arranged in a manner that their hydrophobic tails are inward towards the core, and the hydrophilic heads are at the periphery of the round vesicle to form a micelle with a hydrophobic core and hydrophilic shell. The hydrophobic core helps in the conveying of the hydrophobic drugs and aids in the passage of the hydrophilic drugs across the cell membrane, whereas the hydrophilic shell helps to convey the hydrophilic drugs and aids in the solubilization of micelle in water (Guler et al. 2023). Cationic AMPs have also been found to self-assemble on the hydrophilic surface of micelle, resulting in the reduction of minimum inhibitory concentration (MIC) of those AMPs, thereby enhancing their antimicrobial efficacy (Maleki Dizaj et al. 2022b; Zhou et al. 2016).
Nanoparticles
Nanoparticles (NPs) of different sizes, existing in various forms like metallic NPs, polymeric NPs, and mesoporous silica NPs, have also been utilized in the formulation of multiple AMPs to enhance their efficacy, stability, and specificity against pathogenic species (Carratalá et al. 2020).
Metallic Nanoparticles
Metallic NPs derived from silver and gold have widely been utilized to deliver the AMPs to their target pathogen. These types of NPs have also been associated with their antimicrobial properties (Engin & Engin 2019). For example, silver nanoparticles (AgNPs) have been found to bind and disrupt the negatively charged microbial membrane and, hence, can produce a synergistic effect in conjugation with AMPs (Bruna et al. 2021). A study by Mohanty et al. on developing AMPs-AgNPs conjugates by green synthesis method, utilizing plant extract, had shown a potent bactericidal effect of this novel conjugate against mycobacteria with highly reduced cytotoxicity (Mohanty et al. 2013). Similar to AgNPs, gold nanoparticles (AuNPs) have also shown broad-spectrum antimicrobial activity against various water-borne pathogens, including E. coli, S. Typhimurium, and Shigella dysenteriae and their AMP conjugates had displayed a significant increase in antimicrobial activity (Hammami et al. 2021). A recent study that showed the conjugation of AuNPs with esculentin-1a, an AMP derived from frog skin, had found an increase in the antimicrobial efficacy by 15 folds, compared to esculentin-1a, against P. aeruginosa, along with enhanced proteolytic stability (Casciaro et al. 2017). As most of the nanoparticles can concentrate within the liver, spleen, and lymph nodes, the use of noble metals like silver and gold, which are resistant to oxidation and erosion, is highly preferred for the synthesis of metallic NPs (Maleki Dizaj et al. 2022b).
Polymeric Nanoparticles
Several polymers, like chitosan and its derivatives, PLGA, poly lactic acid (PLA), and poly acrylic acid (PAA), have been utilized to synthesize NPs that have aided in the delivery of multiple drugs to enhance their efficacy and metabolic stability. Various studies have also shown the utilization of polymeric nanoparticles in assisting the delivery of AMPs to increase their bioavailability and overcome other limitations, as they have been found to protect AMPs from proteolytic degradation and enhance drug penetration within the targeted cell by binding to cell membranes (Maleki Dizaj et al. 2022b). A recent study by Almaaytah et al. showed the encapsulation of AMPs in chitosan nanoparticles, which resulted in the reduction of toxicity and enhancement of antimicrobial efficacy of AMP-chitosan nanoparticles, as compared to free AMP (Almaaytah et al. 2017). Another latest study showed the encapsulation of rifampicin in the nanoparticles of the N-acetylcysteine derivative of chitosan, which were further conjugated with the AMP Ctx(Ile21)-Ha, resulting in the synergistic effect of both the drugs with enhanced stability against multi-and extensively-drug resistant Mycobacterium tuberculosis (Primo et al. 2024). The novel star-shaped peptide polymers have recently gained significant importance in biomedical applications and have also been extensively used to synthesize nanoparticles for the encapsulation of AMPs. These star-shaped polymers are also found to be associated with their antimicrobial activity. They can act through various pathways to inhibit microorganisms and reduce host toxicity, thus rendering these nanocarriers more preferential than conventional nanocarriers (Wu et al. 2015).
Novel Inorganic Nanoparticles
The recent developments have also contributed to the synthesis of inorganic nanoparticles. The inorganic nanoparticles have therapeutic attention because of their porous nature, and the recent study by Braun et al. showed that the porosity of these inorganic nanoparticles can control the drug release and loading, thereby achieving sustained drug release and highly controlled drug loading. One such mostly used inorganic nanoparticles are mesoporous silica nanoparticles (MSNs) due to their multiple advantages, including biocompatibility, uniform pore sizes, high pore volume, modifiable particle sizes, and high surface area (Braun et al. 2016; Luo et al. 2023b). However, the utilization of inorganic nanoparticles has been associated with erythrocyte cytotoxicity. However, this can also be lowered using several strategies, such as bonding/capping the cytotoxic group on the nanoparticles (de la Torre et al. 2014; Deslouches et al. 2020).
Nanofibers
Several polymers, like PLA and chitosan, have been utilized in the production of filaments by special processing methods like electrospinning. This results in the synthesis of nanofibers, which are promising nanocarriers for AMPs. Due to their filamentous structure, nanofibers have a high surface-to-volume ratio, thereby enabling them to entrap more AMPs within their vesicles (Hu et al. 2014; Maleki Dizaj et al. 2022b).
Dendrimers
Dendrimers are different nano-delivery systems that consist of well-defined structures with a high degree of molecular uniformity and low polydispersity, rendering them a more preferred material in the development of nanomedicines (Nordström et al. 2019). Dendrimers are developed from several polymers, including poly-amidoamine (PAMAM), poly-propylene imine (PPI), poly-L-lysine (PLL), polyglycerol (PG), poly-benzyl ether, and carbosilane (Kawano et al. 2020). In a recent study by Fernandez et al., the development of the nanoconjugate of carbosilane dendrimer and AMP-containing cysteine derivatives could attach to a maleimide group in the focal point of the dendrimer. The resulting dendrimer-AMP nanoconjugate had been found to penetrate the cell membrane more effectively and had resulted in increased antimicrobial efficacy, as compared to the free AMP molecule (Fernandez et al. 2019).
Carbon Nanotubes
Carbon nanotubes are nano-sized round and hollow cylindrical tubes composed of rolling graphene sheets, in which the bondings between the carbon atoms are unsymmetric. Due to their higher strength and inelastic behaviour, carbon nanotubes have a great application in nanomedicines, aiding in the delivery of drugs that are sensitive to degradation by digestive enzymes, including AMPs (Dizaj et al. 2015; Rathinavel et al. 2021). However, the utilization of carbon nanotubes has been associated with aqueous insolubility, but this was overcome by their functionalization, especially with AMPs (Dizaj et al. 2015).
Hydrogels
Hydrogel is another form of nanocarrier system that is water insoluble and consists of porous 3D matrices, which can absorb water, maintain oxygen penetration, and controllably release drugs, rendering it the preferential drug-delivery system in topical applications. Hydrogels have also been found to be very biocompatible and can carry the AMPs for longer durations against various dermal infections (Borro et al. 2020; Tang et al. 2014). Multiple methods, including the self-assembling octapeptide method and light-induced crosslinking method, have been employed to synthesize the AMP-loaded hydrogels (Tang et al. 2014).
Strategies Modulating Gastrointestinal Tract
GIT’s pH modulation has also enhanced the oral bioavailability of AMPs. As most of the peptides are stable and can be spared from degradation by digestive proteases at a pH range of 6–7, various approaches have been employed to regulate the pH range of the GIT within 6–7 (Koziolek et al. 2015; Zhu et al. 2021). Citric acid has been utilized in association with enteric-coated capsules to maintain the intestinal pH and circumvent the degradation of the peptides in the stomach and the intestine for their effective absorption (Sedyakina et al. 2020). Another strategy of co-administration of AMPs with protease inhibitors has also been employed to enhance their oral bioavailability. Chemical entities like Di-isopropyl fluorophosphate, cholic acids, and their derivatives have been found to inhibit the proteases. Still, they are rarely utilized in the treatment because of their high toxicity (Liu et al. 2018). So, alternative forms of protease inhibitors have been used that consist of modified peptides such as ovomucoids and aprotinin (inhibiting trypsin and chymotrypsin) and soybean trypsin (inhibiting pancreatic endopeptidases) (Qin et al. 2023). Though protease inhibitors have been associated with toxicity, resulting in the deficiency of these enzymes due to their higher doses and long activity, they have still been utilized extensively in the clinical applications of various AMPs (Liu et al. 2018). Several other strategies, including the utilization of absorption enhancers, mucoadhesive and mucus-penetrating systems, enteric-coated and colon-specific delivery, and intestinal microneedles, to enhance the oral bioavailability of peptide drugs have been mentioned in the review study by Quangang Zhu et al. (Zhu et al. 2021).
Antimicrobial Peptides in Clinical Trials
Despite their potential as therapeutic agents, only a limited number of AMPs have successfully advanced to clinical application, primarily due to challenges such as stability, toxicity, and the development of resistance. Recent advancements in biotechnology and bioinformatics have reignited interest in AMPs, leading to the development of synthetic and modified peptides with enhanced efficacy and reduced toxicity. Current clinical trials are exploring the therapeutic potential of these novel AMPs for a variety of indications, from combating resistant bacterial infections to preventing dental caries and treating fungal infections. Table 2 provides an overview of the most promising AMPs currently undergoing clinical trials, highlighting their mechanisms of action, clinical phases, and potential impact on global health.
Future Perspectives and Conclusion
AMPs are an important part of the innate immune system within the host that aids in preventing the colony formation of pathogenic microorganisms and protecting the host from their infections. AMPs can be considered a potent alternative to existing antibiotics in the treatment of infections caused by various MDR pathogenic species, especially ESKAPE pathogens, due to their ability to act through multiple modes of action, which renders them to escape from the development of resistance in these species. With the sudden rise in the development of resistance against existing antibiotics, the urgency to develop an alternative treatment against the MDR pathogenic microorganisms has significantly increased, and certain considerations have been implemented by regulatory authorities to eliminate the regulatory obstacles in the development of novel antimicrobial agents, which potentially contributed to an increase in clinical applications of AMPs. As stated above, some AMPs are also associated with other therapeutic properties like immunomodulatory, anticancer, anti-inflammatory, wound-healing and tissue repair properties. In addition, AMPs have also been found to maintain homeostasis in the intestinal tract by protecting the normal gut microbiota and eliminating the colony formation of pathogenic species within the gut. Therefore, these wide ranges of therapeutic properties will contribute to the diversified clinical applications of AMPs in the upcoming years.
Though AMPs may be considered to be more efficient therapeutically compared to existing antimicrobial agents against various pathogenic species, certain limitations, including the slow development of resistance, that limit the clinical applications of AMPs cannot be ignored. So, at present, the complete replacement of existing antibiotics with the AMPs may not be considerable. However, some of the possible solutions/strategies stated in this article will be helpful to overcome a few limitations associated with AMPs, which may help to direct the current status of AMPs to potential antimicrobial agents in the future. Recent studies have also shown the utilization of more than one strategy to develop potential antimicrobial formulations. One such recent study utilizing more than one strategy to develop potential AMP formulation was performed by Primo et al., which showed the utilization of nanoparticle encapsulation strategy and combination therapy with existing antibiotics. This study included the encapsulation of rifampicin in the nanoparticles of the N-acetylcysteine derivative of chitosan, which were further conjugated with the AMP Ctx(Ile21)-Ha, resulting in the synergistic effect of both the drugs with enhanced stability against multi- and extensively-drug resistant Mycobacterium tuberculosis (Primo et al. 2024). Several local delivery formulations, including sprays, inhalers, ointments, creams, eye drops, films, aerosols, powders, gels, tablets, and patches, have also been developed and utilized clinically for achieving site-specific delivery of AMPs through nasal, topical, ocular, buccal, pulmonary, sublingual, transdermal, vaginal and rectal routes of administration (Erak et al. 2018; Wang et al. 2022).
Regarding synthesis, recent advancements in the production of AMPs, involving automated chemical synthesis and the use of heterologous expression systems in fermentation, have been widely employed for the large-scale production of AMPs industrially. Recent reviews on the development of peptide synthesis by Martin et al. and Isidro-Llobet et al. have stated some currently employed novel strategies for the development of chemical synthesis methods, along with certain modifications in their methodologies to optimize the synthesis of peptides(Isidro-Llobet et al. 2019; Martin et al. 2020). The design-based synthesis methods, involving the study of structure–activity relationships of AMPs and designing the novel peptide sequences with optimized characteristics to synthesize potent, safe, specific, and efficient AMPs, have also gained significant importance in the design and development of AMPs. The availability of various modern tools and techniques, including several bioinformatics software, databases, omics strategies, gene editing tools like CRISPR-Cas9, rDNA technology, machine learning, and artificial intelligence will make a promising future for peptide-based drug developments.
Many recent studies have highlighted the current status of AMPs. Among these studies, one of the recent studies by Chen et al. has also included a summary of all therapeutic peptides approved by the FDA until 2019 (Chen & Lu 2020). This data would further help in the approval processes of novel AMPs for various clinical applications. However, more development and extensive research are needed to synthesize AMPs with ideal characteristics cost-effectively and to overcome all their limitations. Increased research studies for the development and optimization of AMPs, particularly focusing on novel strategies like various combination strategies, nano-formulations, hybridization, etc. and utilizing modern computational tools and techniques, are further needed that can result in the development of potential alternatives to the existing antibiotics and eradicate the deadly diseases caused by MDR pathogenic species.
References
Ageitos JM, Sánchez-Pérez A, Calo-Mata P, Villa TG (2017) Antimicrobial peptides (AMPs): ancient compounds that represent novel weapons in the fight against bacteria. Biochem Pharmacol. https://doi.org/10.1016/j.bcp.2016.09.018
Albada B, Metzler-Nolte N (2017) Highly potent antibacterial organometallic peptide conjugates. Acc Chem Res 50(10):2510. https://doi.org/10.1021/acs.accounts.7b00282
Allahou LW, Madani SY, Seifalian A (2021) Investigating the application of liposomes as drug delivery systems for the diagnosis and treatment of cancer. Int J Biomater 2021:1–16. https://doi.org/10.1155/2021/3041969
Almaaytah A, Mohammed G, Abualhaijaa A, Al-Balas Q (2017) Development of novel ultrashort antimicrobial peptide nanoparticles with potent antimicrobial and antibiofilm activities against multidrug-resistant bacteria. Drug Des Dev Ther 11:3159–3170. https://doi.org/10.2147/DDDT.S147450
Almaaytah A, Qaoud MT, Abualhaijaa A, Al-Balas Q, Alzoubi KH (2018) Hybridization and antibiotic synergism as a tool for reducing the cytotoxicity of antimicrobial peptides. Infect Drug Resist. https://doi.org/10.2147/IDR.S166236
Lewies A, Du Plessis LH, Wentzel JF (2019) Antimicrobial peptides: the Achilles’ heel of antibiotic resistance? Probiotics Antimicrob Proteins 11(2):370–381
Arias M, Piga K, Hyndman M, Vogel H (2018) Improving the activity of trp-rich antimicrobial peptides by Arg/Lys substitutions and changing the length of cationic residues. Biomolecules 8(2):19. https://doi.org/10.3390/biom8020019
Assoni L, Milani B, Carvalho MR, Nepomuceno LN, Waz NT, Guerra MES, Converso TR, Darrieux M (2020) Resistance mechanisms to antimicrobial peptides in gram-positive bacteria. Front Microbiol. https://doi.org/10.3389/fmicb.2020.593215
Bahar A, Ren D (2013) Antimicrobial peptides. Pharmaceuticals 6(12):1543–1575. https://doi.org/10.3390/ph6121543
Bahnsen JS, Franzyk H, Sayers EJ, Jones AT, Nielsen HM (2015) Cell-Penetrating antimicrobial peptides—prospectives for targeting intracellular infections. Pharm Res 32(5):1546–1556. https://doi.org/10.1007/s11095-014-1550-9
Baker JL, He X, Shi W (2019) Precision reengineering of the oral microbiome for caries management. Adv Dent Res. https://doi.org/10.1177/0022034519877386
Ballantine RD, McCallion CE, Nassour E, Tokajian S, Cochrane SA (2019) Tridecaptin-inspired antimicrobial peptides with activity against multidrug-resistant Gram-negative bacteria. MedChemComm 10(3):484–487. https://doi.org/10.1039/C9MD00031C
Band V, Weiss D (2014) Mechanisms of antimicrobial peptide resistance in gram-negative bacteria. Antibiotics 4(1):18–41. https://doi.org/10.3390/antibiotics4010018
Bann SJ, Ballantine RD, Cochrane SA (2021) The tridecaptins: non-ribosomal peptides that selectively target Gram-negative bacteria. RSC Med Chem 12(4):538–551. https://doi.org/10.1039/D0MD00413H
Bechinger B, Gorr SU (2017) Antimicrobial peptides: mechanisms of action and resistance. J Dent Res 96:254–260
Behrendt R, White P, Offer J (2016) Advances in Fmoc solid-phase peptide synthesis. J Peptide Sci. https://doi.org/10.1002/psc.2836
Bellotti D, Remelli M (2022) Lights and shadows on the therapeutic use of antimicrobial peptides. Molecules 27(14):4584. https://doi.org/10.3390/molecules27144584
Bhattacharjee R, Nandi A, Sinha A, Kumar H, Mitra D, Mojumdar A, Patel P, Jha E, Mishra S, Rout PK, Panda PK, Suar M, Verma SK (2022) Phage-tail-like bacteriocins as a biomedical platform to counter anti-microbial resistant pathogens. Biomed Pharmacother. https://doi.org/10.1016/j.biopha.2022.113720
Bin Hafeez A, Jiang X, Bergen PJ, Zhu Y (2021) Antimicrobial peptides: an update on classifications and databases. Int J Mol Sci 22(21):11691. https://doi.org/10.3390/ijms222111691
Borro BC, Nordström R, Malmsten M (2020) Microgels and hydrogels as delivery systems for antimicrobial peptides. Colloids Surf B: Biointerfaces. https://doi.org/10.1016/j.colsurfb.2020.110835
Braun K, Pochert A, Lindén M, Davoudi M, Schmidtchen A, Nordström R, Malmsten M (2016) Membrane interactions of mesoporous silica nanoparticles as carriers of antimicrobial peptides. J Colloid Interface Sci 475:161–170. https://doi.org/10.1016/j.jcis.2016.05.002
Brown KL, Poon GFT, Birkenhead D, Pena OM, Falsafi R, Dahlgren C, Karlsson A, Bylund J, Hancock REW, Johnson P (2011) Host defense peptide LL-37 selectively reduces proinflammatory macrophage responses. J Immunol. https://doi.org/10.4049/jimmunol.1002508
Bruna T, Maldonado-Bravo F, Jara P, Caro N (2021) Silver nanoparticles and their antibacterial applications. Int J Mol Sci. https://doi.org/10.3390/ijms22137202
Cameron A, Zaheer R, Adator EH, Barbieri R, Reuter T, McAllister TA (2019) Bacteriocin occurrence and activity in Escherichia coli isolated from bovines and wastewater. Toxins. https://doi.org/10.3390/toxins11080475
Campos ML, De Souza CM, De Oliveira KBS, Dias SC, Franco OL (2018) The role of antimicrobial peptides in plant immunity. J Exp Bot. https://doi.org/10.1093/jxb/ery294
Cao Q, Ge C, Wang X, Harvey PJ, Zhang Z, Ma Y, Wang X, Jia X, Mobli M, Craik DJ, Jiang T, Yang J, Wei Z, Wang Y, Chang S, Yu R (2023) Designing antimicrobial peptides using deep learning and molecular dynamic simulations. Briefings Bioinform. https://doi.org/10.1093/bib/bbad058
Cardoso MH, Orozco RQ, Rezende SB, Rodrigues G, Oshiro KGN, Cândido ES, Franco OL (2020) Computer-aided design of antimicrobial peptides: are we generating effective drug candidates? Front Microbiol. https://doi.org/10.3389/fmicb.2019.03097
Carratalá JV, Serna N, Villaverde A, Vázquez E, Ferrer-Miralles N (2020) Nanostructured antimicrobial peptides: the last push towards clinics. Biotechnol Adv. https://doi.org/10.1016/j.biotechadv.2020.107603
Casciaro B, Moros M, Rivera-Fernández S, Bellelli A, de la Fuente JM, Mangoni ML (2017) Gold-nanoparticles coated with the antimicrobial peptide esculentin-1a(1–21)NH2 as a reliable strategy for antipseudomonal drugs. Acta Biomater 47:170–181. https://doi.org/10.1016/j.actbio.2016.09.041
Casciaro B, d’Angelo I, Zhang X, Loffredo MR, Conte G, Cappiello F, Quaglia F, Di Y-PP, Ungaro F, Mangoni ML (2019) Poly(lactide-co-glycolide) nanoparticles for prolonged therapeutic efficacy of Esculentin-1a-Derived antimicrobial peptides against Pseudomonas aeruginosa lung infection: in vitro and in vivo studies. Biomacromol 20(5):1876–1888. https://doi.org/10.1021/acs.biomac.8b01829
De Cesare GB, Cristy SA, Garsin DA, Lorenz MC (2020) Antimicrobial peptides: a new frontier in antifungal therapy. Mbio. https://doi.org/10.1128/mBio.02123-20
Chegini PP, Nikokar I, Tabarzad M, Faezi S, Mahboubi A (2019) Effect of amino acid substitutions on biological activity of antimicrobial peptide: Design, recombinant production, and biological activity. Iranian J Pharm Res. https://doi.org/10.22037/ijpr.2019.112397.13734
Chen CH, Lu TK (2020) Development and challenges of antimicrobial peptides for therapeutic applications. Antibiotics 9(1):24. https://doi.org/10.3390/antibiotics9010024
Chen L, Shen T, Liu Y, Zhou J, Shi S, Wang Y, Zhao Z, Yan Z, Liao C, Wang C (2020) Enhancing the antibacterial activity of antimicrobial peptide PMAP-37(F34-R) by cholesterol modification. BMC Veterinary Res. https://doi.org/10.1186/s12917-020-02630-x
Choi GH, Holzapfel WH, Todorov SD (2023) Diversity of the bacteriocins, their classification and potential applications in combat of antibiotic resistant and clinically relevant pathogens. Crit Rev Microbiol. https://doi.org/10.1080/1040841X.2022.2090227
Collin F, Maxwell A (2019) The microbial toxin microcin B17: prospects for the development of new antibacterial agents. J Mol Biol. https://doi.org/10.1016/j.jmb.2019.05.050
Conibear AC, Chaousis S, Durek T, Johan Rosengren K, Craik DJ, Schroeder CI (2016) Approaches to the stabilization of bioactive epitopes by grafting and peptide cyclization. Biopolymers. https://doi.org/10.1002/bip.22767
Conlon JM, Sonnevend A (2010) Antimicrobial peptides in frog skin secretions. Methods Mol Biol. https://doi.org/10.1007/978-1-60761-594-1_1
Cui Z, Luo Q, Bannon MS, Gray VP, Bloom TG, Clore MF, Hughes MA, Crawford MA, Letteri RA (2021) Molecular engineering of antimicrobial peptide (AMP)–polymer conjugates. Biomater Sci 9(15):5069–5091. https://doi.org/10.1039/D1BM00423A
Dahiya R, Kumar S, Khokra S, Gupta S, Sutariya V, Bhatia D, Sharma A, Singh S, Maharaj S (2018) Toward the synthesis and improved biopotential of an N-methylated analog of a Proline-Rich cyclic tetrapeptide from marine bacteria. Mar Drugs 16(9):305. https://doi.org/10.3390/md16090305
Das, P., Sercu, T., Wadhawan, K., Padhi, I., Gehrmann, S., Cipcigan, F., Chenthamarakshan, V., Strobelt, H., Santos, C., Chen, P.-Y., Yang, Y. Y., Tan, J., Hedrick, J., Crain, J., & Mojsilovic, A. (2020). Accelerating antimicrobial discovery with controllable deep generative models and molecular dynamics. In arXiv.
Dash R, Bhattacharjya S (2021) Thanatin: An emerging host defense antimicrobial peptide with multiple modes of action. Int J Mol Sci. https://doi.org/10.3390/ijms22041522
David AA, Park SE, Parang K, Tiwari RK (2018) Antibiotics-peptide conjugates against multidrug-resistant bacterial pathogens. Curr Topics Med Chem 18(22):1926–1936. https://doi.org/10.2174/1568026619666181129141524
Deslouches B, Montelaro RC, Urish KL, Di YP (2020) Engineered cationic antimicrobial peptides (eCAPs) to combat multidrug-resistant bacteria. Pharmaceutics 12(6):501. https://doi.org/10.3390/pharmaceutics12060501
Diamond G, Molchanova N, Herlan C, Fortkort JA, Lin JS, Figgins E, Bopp N, Ryan LK, Chung D, Adcock RS, Sherman M, Barron AE (2021) Potent antiviral activity against HSV-1 and SARS-CoV-2 by antimicrobial peptoids. Pharmaceuticals. https://doi.org/10.3390/ph14040304
Dintner S, Staroń A, Berchtold E, Petri T, Mascher T, Gebhard S (2011) Coevolution of ABC transporters and two-component regulatory systems as resistance modules against antimicrobial peptides in firmicutes bacteria. J Bacteriol 193(15):3851–3862. https://doi.org/10.1128/JB.05175-11
Dizaj SM, Mennati A, Jafari S, Khezri K, Adibkia K (2015) Antimicrobial activity of carbon-based nanoparticles. Adv Pharm Bull. https://doi.org/10.5681/apb.2015.003
Drexelius M, Reinhardt A, Grabeck J, Cronenberg T, Nitsche F, Huesgen PF, Maier B, Neundorf I (2021) Multistep optimization of a cell-penetrating peptide towards its antimicrobial activity. Biochem J. https://doi.org/10.1042/BCJ20200698
Duclohier H (2010) Antimicrobial peptides and peptaibols substitutes for conventional antibiotics. Curr Pharm Design. https://doi.org/10.2174/138161210793292500
Eggink D, Bontjer I, de Taeye SW, Langedijk JPM, Berkhout B, Sanders RW (2019) HIV-1 anchor inhibitors and membrane fusion inhibitors target distinct but overlapping steps in virus entry. J Biol Chem. https://doi.org/10.1074/jbc.RA119.007360
Van Eijk M, Boerefijn S, Cen L, Rosa M, Morren MJH, Van Der Ent CK, Kraak B, Dijksterhuis J, Valdes ID, Haagsman HP, De Cock H (2020) Cathelicidin-inspired antimicrobial peptides as novel antifungal compounds. Med Mycol. https://doi.org/10.1093/mmy/myaa014
Engin AB, Engin A (2019) Nanoantibiotics: a novel rational approach to antibiotic resistant infections. Curr Drug Metab. https://doi.org/10.2174/1389200220666190806142835
Erak M, Bellmann-Sickert K, Els-Heindl S, Beck-Sickinger AG (2018) Peptide chemistry toolbox—Transforming natural peptides into peptide therapeutics. Bioorganic Med Chem. https://doi.org/10.1016/j.bmc.2018.01.012
Essig A, Hofmann D, Münch D, Gayathri S, Künzler M, Kallio PT, Sahl HG, Wider G, Schneider T, Aebi M (2014) Copsin, a novel peptide-based fungal antibiotic interfering with the peptidoglycan synthesis. J Biol Chem. https://doi.org/10.1074/jbc.M114.599878
Falciani C, Lozzi L, Scali S, Brunetti J, Bracci L, Pini A (2014) Site-specific pegylation of an antimicrobial peptide increases resistance to Pseudomonas aeruginosa elastase. Amino Acids 46(5):1403–1407. https://doi.org/10.1007/s00726-014-1686-2
Fernandez J, Acosta G, Pulido D, Malý M, Copa-Patiño JL, Soliveri J, Royo M, Gómez R, Albericio F, Ortega P, de la Mata FJ (2019) Carbosilane dendron-peptide nanoconjugates as antimicrobial agents. Mol Pharm 16(6):2661–2674. https://doi.org/10.1021/acs.molpharmaceut.9b00222
Fjell CD, Hiss JA, Hancock REW, Schneider G (2012) Designing antimicrobial peptides: form follows function. Nat Rev Drug Discovery 11(1):37–51. https://doi.org/10.1038/nrd3591
Friedman ND, Temkin E, Carmeli Y (2016) The negative impact of antibiotic resistance. Clin Microbiol Infect 22(5):416–422. https://doi.org/10.1016/j.cmi.2015.12.002
De La Fuente-Núñez C, Reffuveille F, Mansour SC, Reckseidler-Zenteno SL, Hernández D, Brackman G, Coenye T, Hancock REW (2015) D-Enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal pseudomonas aeruginosa infections. Chem Biol. https://doi.org/10.1016/j.chembiol.2015.01.002
Gagnon MG, Roy RN, Lomakin IB, Florin T, Mankin AS, Steitz TA (2016) Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw018
Galván AE, Chalón MC, Ríos Colombo NS, Schurig-Briccio LA, Sosa-Padilla B, Gennis RB, Bellomio A (2019) Microcin J25 inhibits ubiquinol oxidase activity of purified cytochrome bd-I from Escherichia coli. Biochimie. https://doi.org/10.1016/j.biochi.2019.02.007
Gharsallaoui A, Oulahal N, Joly C, Degraeve P (2016) Nisin as a Food Preservative: Part 1: Physicochemical Properties, Antimicrobial Activity, and Main Uses. Crit Rev Food Sci Nutr. https://doi.org/10.1080/10408398.2013.763765
Gottler LM, Ramamoorthy A (2009) Structure, membrane orientation, mechanism, and function of pexiganan - A highly potent antimicrobial peptide designed from magainin. Biochimica Et Biophysica Acta—Biomembranes. https://doi.org/10.1016/j.bbamem.2008.10.009
Graf M, Wilson DN (2019) Intracellular antimicrobial peptides targeting the protein synthesis machinery. Adv Exp Med Biol. https://doi.org/10.1007/978-981-13-3588-4_6
Grassi L, Maisetta G, Esin S, Batoni G (2017) Combination strategies to enhance the efficacy of antimicrobial peptides against bacterial biofilms. Front Microbiol. https://doi.org/10.3389/fmicb.2017.02409
Greco I, Molchanova N, Holmedal E, Jenssen H, Hummel BD, Watts JL, Håkansson J, Hansen PR, Svenson J (2020) Correlation between hemolytic activity, cytotoxicity and systemic in vivo toxicity of synthetic antimicrobial peptides. Scientific Rep. https://doi.org/10.1038/s41598-020-69995-9
Van Groenendael R, Beunders R, Hemelaar P, Hofland J, Morshuis WJ, Van Der Hoeven JG, Gerretsen J, Wensvoort G, Kooistra EJ, Claassen WJ, Waanders D, Lamberts MGA, Buijsse LSE, Kox M, Van Eijk LT, Pickkers P (2021) Safety and efficacy of human chorionic gonadotropin hormone-derivative EA-230 in cardiac surgery patients: a randomized double-blind placebo-controlled study. Crit Care Med. https://doi.org/10.1097/CCM.0000000000004847
Guha S, Ferrie RP, Ghimire J, Ventura CR, Wu E, Sun L, Kim SY, Wiedman GR, Hristova K, Wimley WC (2021) Applications and evolution of melittin, the quintessential membrane active peptide. Biochem Pharmacol. https://doi.org/10.1016/j.bcp.2021.114769
Guler E, Polat EB, Cam ME (2023) Drug delivery systems for neural tissue engineering. Biomater Neural Tissue Eng. https://doi.org/10.1016/B978-0-323-90554-1.00012-4
Gunasekera S, Muhammad T, Strömstedt AA, Rosengren KJ, Göransson U (2020) Backbone cyclization and dimerization of LL-37-Derived peptides enhance antimicrobial activity and proteolytic stability. Front Microbiol. https://doi.org/10.3389/fmicb.2020.00168
Håkansson J, Ringstad L, Umerska A, Johansson J, Andersson T, Boge L, Rozenbaum RT, Sharma PK, Tollbäck P, Björn C, Saulnier P, Mahlapuu M (2019) Characterization of the in vitro, ex vivo, and in vivo efficacy of the antimicrobial peptide DPK-060 used for topical treatment. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2019.00174
Hammami I, Alabdallah NM, Al Jomaa A, Kamoun M (2021) Gold nanoparticles: Synthesis properties and applications. J King Saud Univ—Sci. https://doi.org/10.1016/j.jksus.2021.101560
Haney EF, Mansour SC, Hancock REW (2017) Antimicrobial peptides: an introduction. Antimicrob Peptides: Methods Protoc. https://doi.org/10.1007/978-1-4939-6737-7_1
He M, Zhang H, Li Y, Wang G, Tang B, Zhao J, Huang Y, Zheng J (2018) Cathelicidin-derived antimicrobial peptides inhibit Zika virus through direct inactivation and interferon pathway. Front Immunol. https://doi.org/10.3389/fimmu.2018.00722
Hirano M, Saito C, Yokoo H, Goto C, Kawano R, Misawa T, Demizu Y (2021) Development of antimicrobial stapled peptides based on magainin 2 sequence. Molecules. https://doi.org/10.3390/molecules26020444
Hoelscher MP, Forner J, Calderone S, Krämer C, Taylor Z, Loiacono FV, Agrawal S, Karcher D, Moratti F, Kroop X, Bock R (2022) Expression strategies for the efficient synthesis of antimicrobial peptides in plastids. Nat Commun. https://doi.org/10.1038/s41467-022-33516-1
van Hoek ML (2014) Antimicrobial peptides in reptiles. Pharmaceuticals. https://doi.org/10.3390/ph7060723
Hu X, Liu S, Zhou G, Huang Y, Xie Z, Jing X (2014) Electrospinning of polymeric nanofibers for drug delivery applications. J Control Release 185:12–21. https://doi.org/10.1016/j.jconrel.2014.04.018
Huan Y, Kong Q, Mou H, Yi H (2020) Antimicrobial peptides: classification, design, application and research progress in multiple fields. Front Microbiol. https://doi.org/10.3389/fmicb.2020.582779
Huang DB, Brothers KM, Mandell JB, Taguchi M, Alexander PG, Parker DM, Shinabarger D, Pillar C, Morrissey I, Hawser S, Ghahramani P, Dobbins D, Pachuda N, Montelaro R, Steckbeck JD, Urish KL (2022) Engineered peptide PLG0206 overcomes limitations of a challenging antimicrobial drug class. PLoS ONE. https://doi.org/10.1371/journal.pone.0274815
Hussein M, Karas JA, Schneider-Futschik EK, Chen F, Swarbrick J, Paulin OKA, Hoyer D, Baker M, Zhu Y, Li J, Velkov T (2020) The killing mechanism of teixobactin against methicillin-resistant staphylococcus aureus: an untargeted metabolomics study. Msystems. https://doi.org/10.1128/msystems.00077-20
Isidro-Llobet A, Kenworthy MN, Mukherjee S, Kopach ME, Wegner K, Gallou F, Smith AG, Roschangar F (2019) Sustainability challenges in peptide synthesis and purification: from R&D to production. J Organic Chem. https://doi.org/10.1021/acs.joc.8b03001
Jash A, Ubeyitogullari A, Rizvi SSH (2021) Liposomes for oral delivery of protein and peptide-based therapeutics: challenges, formulation strategies, and advances. J Mater Chem B. https://doi.org/10.1039/d1tb00126d
Ji Z, Li F, Xia Z, Guo X, Gao M, Sun F, Cheng Y, Wu Y, Li W, Ali SA, Cao Z (2018) The scorpion venom peptide Smp76 inhibits viral infection by regulating Type-I interferon response. Virologica Sinica. https://doi.org/10.1007/s12250-018-0068-4
Ji S, An F, Zhang T, Lou M, Guo J, Liu K, Zhu Y, Wu J, Wu R (2024) Antimicrobial peptides: an alternative to traditional antibiotics. Eur J Med Chem 265:116072. https://doi.org/10.1016/j.ejmech.2023.116072
Jolivet-Gougeon A, Bonnaure-Mallet M (2014) Biofilms as a mechanism of bacterial resistance. Drug Discov Today: Technol. https://doi.org/10.1016/j.ddtec.2014.02.003
Joo HS, Fu CI, Otto M (2016) Bacterial strategies of resistance to antimicrobial peptides. Phil Trans Royal Soc b: Biol Sci. https://doi.org/10.1098/rstb.2015.0292
Kawano Y, Jordan O, Hanawa T, Borchard G, Patrulea V (2020) Are antimicrobial peptide dendrimers an escape from ESKAPE? Adv Wound Care 9(7):378–395. https://doi.org/10.1089/wound.2019.1113
Khademi M, Varasteh-Shams M, Nazarian-Firouzabadi F, Ismaili A (2020) New recombinant antimicrobial peptides confer resistance to fungal pathogens in tobacco plants. Front Plant Sci. https://doi.org/10.3389/fpls.2020.01236
Kim GC, Cheon DH, Lee Y (2021) Challenge to overcome current limitations of cell-penetrating peptides. Biochimica Et Biophysica Acta—Proteins Proteomics. https://doi.org/10.1016/j.bbapap.2021.140604
Knappe D, Goldbach T, Hatfield M, Palermo N, Weinert S, Sträter N, Hoffmann R, Lovas S (2016) Proline-rich antimicrobial peptides optimized for binding to Escherichia coli Chaperone DnaK. Protein Peptide Lett. https://doi.org/10.2174/0929866523666160719124712
Kovács R, Nagy F, Tóth Z, Bozó A, Balázs B, Majoros L (2019) Synergistic effect of nikkomycin Z with caspofungin and micafungin against Candida albicans and Candida parapsilosis biofilms. Lett Appl Microbiol. https://doi.org/10.1111/lam.13204
Koziolek M, Grimm M, Becker D, Iordanov V, Zou H, Shimizu J, Wanke C, Garbacz G, Weitschies W (2015) Investigation of pH and temperature profiles in the GI tract of fasted human subjects using the Intellicap® system. J Pharm Sci. https://doi.org/10.1002/jps.24274
Latka A, Maciejewska B, Majkowska-Skrobek G, Briers Y, Drulis-Kawa Z (2017) Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-017-8224-6
Lau JL, Dunn MK (2018) Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic Med Chem. https://doi.org/10.1016/j.bmc.2017.06.052
Le CF, Fang CM, Sekaran SD (2017) Intracellular targeting mechanisms by antimicrobial peptides. Antimicrobial Agents Chemother. https://doi.org/10.1128/AAC.02340-16
Lee H, Lim SI, Shin SH, Lim Y, Koh JW, Yang S (2019) Conjugation of cell-penetrating peptides to antimicrobial peptides enhances antibacterial activity. ACS Omega. https://doi.org/10.1021/acsomega.9b02278
Li T, Li L, Du F, Sun L, Shi J, Long M, Chen Z (2021) Activity and mechanism of action of antifungal peptides from microorganisms: a review. Molecules. https://doi.org/10.3390/molecules26113438
Li X, Zuo S, Wang B, Zhang K, Wang Y (2022) Antimicrobial mechanisms and clinical application prospects of antimicrobial peptides. Molecules 27(9):2675. https://doi.org/10.3390/molecules27092675
Li T, Wang Z, Guo J, de la Fuente-Nunez C, Wang J, Han B, Tao H, Liu J, Wang X (2023) Bacterial resistance to antibacterial agents: Mechanisms, control strategies, and implications for global health. Sci Total Environ. https://doi.org/10.1016/j.scitotenv.2022.160461
Lima PG, Oliveira JTA, Amaral JL, Freitas CDT, Souza PFN (2021) Synthetic antimicrobial peptides: Characteristics, design, and potential as alternative molecules to overcome microbial resistance. Life Sci. https://doi.org/10.1016/j.lfs.2021.119647
Lin TT, Yang LY, Lin CY, Wang CT, Lai CW, Ko CF, Shih YH, Chen SH (2023) Intelligent de novo design of novel antimicrobial peptides against antibiotic-resistant bacteria strains. Int J Mol Sci. https://doi.org/10.3390/ijms24076788
Liu C, Kou Y, Zhang X, Cheng H, Chen X, Mao S (2018) Strategies and industrial perspectives to improve oral absorption of biological macromolecules. Expert Opin Drug Deliv. https://doi.org/10.1080/17425247.2017.1395853
Liu Y, Li S, Shen T, Chen L, Zhou J, Shi S, Wang Y, Zhao Z, Liao C, Wang C (2020) N-terminal myristoylation enhanced the antimicrobial activity of antimicrobial peptide PMAP-36PW. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2020.00450
Lu J, Xu H, Xia J, Ma J, Xu J, Li Y, Feng J (2020) D-and unnatural amino acid substituted antimicrobial peptides with improved proteolytic resistance and their proteolytic degradation characteristics. Front Microbiol. https://doi.org/10.3389/fmicb.2020.563030
Luo Y, Song Y (2021) Mechanism of antimicrobial peptides: antimicrobial, anti-inflammatory and antibiofilm activities. Int Mol Sci. https://doi.org/10.3390/ijms222111401
Luo X, Zhu W, Ding L, Ye X, Gao H, Tai X, Wu Z, Qian Y, Ruan X, Li J, Li S, Chen Z (2019) Bldesin, the first functionally characterized pathogenic fungus defensin with Kv1.3 channel and chymotrypsin inhibitory activities. J Biochem Mol Toxicol. https://doi.org/10.1002/jbt.22244
Luo X, Chen H, Song Y, Qin Z, Xu L, He N, Tan Y, Dessie W (2022) Advancements, challenges and future perspectives on peptide-based drugs: focus on antimicrobial peptides. Eur J Pharm Sci 181:106363
Luo X, Chen H, Song Y, Qin Z, Xu L, He N, Tan Y, Dessie W (2023a) Advancements, challenges and future perspectives on peptide-based drugs: Focus on antimicrobial peptides. Eur J Pharma Sci. https://doi.org/10.1016/j.ejps.2022.106363
Luo X, Chen H, Song Y, Qin Z, Xu L, He N, Tan Y, Dessie W (2023b) Advancements, challenges and future perspectives on peptide-based drugs: focus on antimicrobial peptides. Eur J Pharm Sci 181:106363. https://doi.org/10.1016/j.ejps.2022.106363
Ma H, Zhao X, Yang L, Su P, Fu P, Peng J, Yang N, Guo G (2020) Antimicrobial peptide AMP-17 affects candida albicans by disrupting its cell wall and cell membrane integrity. Infect Drug Res. https://doi.org/10.2147/IDR.S250278
Mahlapuu M, Björn C, Ekblom J (2020) Antimicrobial peptides as therapeutic agents: opportunities and challenges. Crit Rev Biotechnol. https://doi.org/10.1080/07388551.2020.1796576
Maiti BK (2020) Potential role of peptide-based antiviral therapy against SARS-CoV-2 infection. ACS Pharmacol Transl Sci. https://doi.org/10.1021/acsptsci.0c00081
Malanovic N, Lohner K (2016) Antimicrobial peptides targeting Gram-positive bacteria. Pharmaceuticals. https://doi.org/10.3390/ph9030059
Maleki Dizaj S, Salatin S, Khezri K, Lee J-Y, Lotfipour F (2022a) Targeting multidrug resistance with antimicrobial peptide-decorated nanoparticles and polymers. Front Microbiol. https://doi.org/10.3389/fmicb.2022.831655
Martin V, Egelund PHG, Johansson H, Thordal Le Quement S, Wojcik F, Sejer Pedersen D (2020) Greening the synthesis of peptide therapeutics: an industrial perspective. RSC Adv. https://doi.org/10.1039/d0ra07204d
Mercer DK, Robertson JC, Miller L, Stewart CS, O’Neil DA (2020) NP213 (Novexatin®): a unique therapy candidate for onychomycosis with a differentiated safety and efficacy profile. Med Mycol. https://doi.org/10.1093/mmy/myaa015
Mohanty S, Jena P, Mehta R, Pati R, Banerjee B, Patil S, Sonawane A (2013) cationic antimicrobial peptides and biogenic silver nanoparticles kill mycobacteria without eliciting DNA damage and cytotoxicity in mouse macrophages. Antimicrob Agents Chemother 57(8):3688–3698. https://doi.org/10.1128/AAC.02475-12
Moiola M, Memeo MG, Quadrelli P (2019) Stapled peptides-a useful improvement for peptide-based drugs. Molecules. https://doi.org/10.3390/molecules24203654
Moretta A, Scieuzo C, Petrone AM, Salvia R, Manniello MD, Franco A, Lucchetti D, Vassallo A, Vogel H, Sgambato A, Falabella P (2021a) Antimicrobial peptides: a new hope in biomedical and pharmaceutical fields. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2021.668632
Mukhopadhyay S, Bharath Prasad AS, Mehta CH, Nayak UY (2020) Antimicrobial peptide polymers: no escape to ESKAPE pathogens—a review. World J Microbiol Biotechnol 36(9):131. https://doi.org/10.1007/s11274-020-02907-1
Münch D, Engels I, Müller A, Reder-Christ K, Falkenstein-Paul H, Bierbaum G, Grein F, Bendas G, Sahl HG, Schneider T (2015) Structural variations of the cell wall precursor lipid II and their influence on binding and activity of the lipoglycopeptide antibiotic oritavancin. Antimicrobial Agents Chemother. https://doi.org/10.1128/AAC.02663-14
Mura M, Wang J, Zhou Y, Pinna M, Zvelindovsky AV, Dennison SR, Phoenix DA (2016) The effect of amidation on the behaviour of antimicrobial peptides. Eur Biophys J 45(3):195–207. https://doi.org/10.1007/s00249-015-1094-x
Mwangi J, Yin Y, Wang G, Yang M, Li Y, Zhang Z, Lai R (2019) The antimicrobial peptide ZY4 combats multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii infection. Proc Natl Acad Sci 116(52):26516–26522. https://doi.org/10.1073/pnas.1909585117
Mylonakis E, Podsiadlowski L, Muhammed M, Vilcinskas A (2016) Diversity, evolution and medical applications of insect antimicrobial peptides. Philos Transact Royal Soc b: Biol Sci. https://doi.org/10.1098/rstb.2015.0290
Nakamura I, Ohsumi K, Takeda S, Katsumata K, Matsumoto S, Akamatsu S, Mitori H, Nakai T (2019) ASp2397 is a novel natural compound that exhibits rapid and potent fungicidal activity against Aspergillus species through a specific transporter. Antimicrobial Agents Chemother. https://doi.org/10.1128/AAC.02689-18
Nath S, Buell AK, Barz B (2023) Pyroglutamate-modified amyloid β(3–42) monomer has more β-sheet content than the amyloid β(1–42) monomer. Phys Chem Chem Phys. https://doi.org/10.1039/d2cp05961d
Nawrot R, Barylski J, Nowicki G, Broniarczyk J, Buchwald W, Goździcka-Józefiak A (2014) Plant antimicrobial peptides. Folia Microbiol 59(3):181–196. https://doi.org/10.1007/s12223-013-0280-4
O’Neill, J. (2016). Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. Government of the United Kingdom. .
O’Neill, J. T. (2016). Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
Nordström R, Malmsten M (2017) Delivery systems for antimicrobial peptides. Adv Colloid Interface Sci. https://doi.org/10.1016/j.cis.2017.01.005
Nordström R, Andrén OCJ, Singh S, Malkoch M, Davoudi M, Schmidtchen A, Malmsten M (2019) Degradable dendritic nanogels as carriers for antimicrobial peptides. J Colloid Interface Sci. https://doi.org/10.1016/j.jcis.2019.07.028
Nuti N, Rottmann P, Stucki A, Koch P, Panke S, Dittrich PS (2022) A multiplexed cell-free assay to screen for antimicrobial peptides in double emulsion droplets. Angew Chem—Int Ed. https://doi.org/10.1002/anie.202114632
Oliva R, Chino M, Pane K, Pistorio V, De Santis A, Pizzo E, D’Errico G, Pavone V, Lombardi A, Del Vecchio P, Notomista E, Nastri F, Petraccone L (2018) Exploring the role of unnatural amino acids in antimicrobial peptides. Sci Rep 8(1):8888. https://doi.org/10.1038/s41598-018-27231-5
Pahar B, Madonna S, Das A, Albanesi C, Girolomoni G (2020) Immunomodulatory role of the antimicrobial ll-37 peptide in autoimmune diseases and viral infections. Vaccines. https://doi.org/10.3390/vaccines8030517
Pan Y-J, Lin T-L, Chen C-C, Tsai Y-T, Cheng Y-H, Chen Y-Y, Hsieh P-F, Lin Y-T, Wang J-T (2017) Klebsiella phage ΦK64–1 encodes multiple depolymerases for multiple host capsular types. J Virol. https://doi.org/10.1128/JVI.02457-16
Pan CY, Tsai TY, Su BC, Hui CF, Chen JY (2017) Study of the antimicrobial activity of tilapia piscidin 3 (TP3) and TP4 and their effects on immune functions in hybrid tilapia (Oreochromis spp.). PLoS ONE. https://doi.org/10.1371/journal.pone.0169678
Pandi A, Adam D, Zare A, Trinh VT, Schaefer SL, Burt M, Klabunde B, Bobkova E, Kushwaha M, Foroughijabbari Y, Braun P, Spahn C, Preußer C, Pogge von Strandmann E, Bode HB, von Buttlar H, Bertrams W, Jung AL, Abendroth F, Erb TJ (2023) Cell-free biosynthesis combined with deep learning accelerates de novo-development of antimicrobial peptides. Nat Commun. https://doi.org/10.1038/s41467-023-42434-9
Peng SY, You RI, Lai MJ, Lin NT, Chen LK, Chang KC (2017) Highly potent antimicrobial modified peptides derived from the Acinetobacter baumannii phage endolysin LysAB2. Scientific Rep. https://doi.org/10.1038/s41598-017-11832-7
Primo LMDG, Roque-Borda CA, Carnero Canales CS, Caruso IP, de Lourenço IO, Colturato VMM, Sábio RM, de Melo FA, Vicente EF, Chorilli M, da Silva Barud H, Barbugli PA, Franzyk H, Hansen PR, Pavan FR (2024) Antimicrobial peptides grafted onto the surface of N-acetylcysteine-chitosan nanoparticles can revitalize drugs against clinical isolates of Mycobacterium tuberculosis. Carbohydr Polym. https://doi.org/10.1016/j.carbpol.2023.121449
Prince A, Sandhu P, Kumar P, Dash E, Sharma S, Arakha M, Jha S, Akhter Y, Saleem M (2016) Lipid-II independent antimicrobial mechanism of nisin depends on its crowding and degree of oligomerization. Scientific Rep. https://doi.org/10.1038/srep37908
Purwin M, Markowska A, Bruzgo I, Rusak T, Surażyński A, Jaworowska U, Midura-Nowaczek K (2017) Peptides with 6-aminohexanoic acid: synthesis and evaluation as plasmin inhibitors. Int J Pept Res Ther 23(2):235–245. https://doi.org/10.1007/s10989-016-9555-3
Qin L, Cui Z, Wu Y, Wang H, Zhang X, Guan J, Mao S (2023) Challenges and strategies to enhance the systemic absorption of inhaled peptides and proteins. Pharm Res. https://doi.org/10.1007/s11095-022-03435-3
Qvit N, Rubin SJS, Urban TJ, Mochly-Rosen D, Gross ER (2017) Peptidomimetic therapeutics: scientific approaches and opportunities. Drug Discov Today. https://doi.org/10.1016/j.drudis.2016.11.003
Rahman MU, Wang W, Sun Q, Shah JA, Li C, Sun Y, Li Y, Zhang B, Chen W, Wang S (2021) Endolysin, a promising solution against antimicrobial resistance. Antibiotics. https://doi.org/10.3390/antibiotics10111277
Ramachander Turaga VN (2020) Peptaibols: Antimicrobial peptides from fungi. Bioact Nat Prod Drug Discov. https://doi.org/10.1007/978-981-15-1394-7_26
Ramamourthy G, Park J, Seo C, Vogel HJ, Park Y (2020) Antifungal and antibiofilm activities and the mechanism of action of repeating lysine-tryptophan peptides against Candida albicans. Microorganisms. https://doi.org/10.3390/microorganisms8050758
Rathinavel S, Priyadharshini K, Panda D (2021) A review on carbon nanotube: an overview of synthesis, properties, functionalization, characterization, and the application. Mater Sci Eng, B 268:115095. https://doi.org/10.1016/j.mseb.2021.115095
Ree R, Varland S, Arnesen T (2018) Spotlight on protein N-terminal acetylation. Exp Mol Med 50(7):1–13. https://doi.org/10.1038/s12276-018-0116-z
Renukuntla J, Vadlapudi AD, Patel A, Boddu SHS, Mitra AK (2013) Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm. https://doi.org/10.1016/j.ijpharm.2013.02.030
Rijsbergen M, Rijneveld R, Todd M, Feiss GL, Kouwenhoven STP, Quint KD, van Alewijk DCJG, de Koning MNC, Klaassen ES, Burggraaf J, Rissmann R, van Poelgeest MIE (2020) Results of phase 2 trials exploring the safety and efficacy of omiganan in patients with human papillomavirus-induced genital lesions. Br J Clin Pharmacol. https://doi.org/10.1111/bcp.14181
Rima M, Rima M, Fajloun Z, Sabatier J-M, Bechinger B, Naas T (2021) Antimicrobial peptides: a potent alternative to antibiotics. Antibiotics 10(9):1095. https://doi.org/10.3390/antibiotics10091095
Rojas-Pirela M, Kemmerling U, Quiñones W, Michels PAM, Rojas V (2023) Antimicrobial peptides (AMPs): potential therapeutic strategy against trypanosomiases? Biomolecules. https://doi.org/10.3390/biom13040599
Rokitskaya TI, Kolodkin NI, Kotova EA, Antonenko YN (2011) Indolicidin action on membrane permeability: Carrier mechanism versus pore formation. Biochimica Et Biophysica Acta—Biomembr. https://doi.org/10.1016/j.bbamem.2010.09.005
Ryu W-S (2017) Virus life cycle. Mol Virol Hum Pathog Viruses. https://doi.org/10.1016/b978-0-12-800838-6.00003-5
Sader HS, Dale GE, Rhomberg PR, Flamm RK (2018) Antimicrobial activity of murepavadin tested against clinical isolates of pseudomonas aeruginosa from the United States, Europe, and China. Antimicrobial Agents Chemother. https://doi.org/10.1128/AAC.00311-18
Saikia K, Sravani YD, Ramakrishnan V, Chaudhary N (2017) Highly potent antimicrobial peptides from N-terminal membrane-binding region of E. coli MreB. Sci Rep 7(1):42994. https://doi.org/10.1038/srep42994
Schnaider L, Rosenberg A, Kreiser T, Kolusheva S, Gazit E, Berman J (2020) Peptide self-assembly is linked to antibacterial, but not antifungal, activity of histatin 5 derivatives. Msphere. https://doi.org/10.1128/msphere.00021-20
Scholl D (2017) Phage tail-like bacteriocins. Ann Rev Virol. https://doi.org/10.1146/annurev-virology-101416-041632
Scudiero O, Nigro E, Cantisani M, Colavita I, Leone M, Mercurio FA, Galdiero M, Pessi A, Daniele A, Salvatore F, Galdiero S (2015) Design and activity of a cyclic mini-β-defensin analog: a novel antimicrobial tool. Int J Nanomed. https://doi.org/10.2147/IJN.S89610
Sedyakina N, Kuskov A, Velonia K, Feldman N, Lutsenko S, Avramenko G (2020) Modulation of entrapment efficiency and in vitro release properties of BSA-loaded chitosan microparticles cross-linked with citric acid as a potential protein-drug delivery system. Materials. https://doi.org/10.3390/MA13081989
Shafee TMA, Lay FT, Phan TK, Anderson MA, Hulett MD (2017) Convergent evolution of defensin sequence, structure and function. Cell Mol Life Sci. https://doi.org/10.1007/s00018-016-2344-5
Shagaghi N, Palombo EA, Clayton AHA, Bhave M (2016) Archetypal tryptophan-rich antimicrobial peptides: properties and applications. World J Microbiol Biotechnol. https://doi.org/10.1007/s11274-015-1986-z
Shah NR, Hancock REW, Fernandez RC (2014) Bordetella pertussis lipid a glucosamine modification confers resistance to cationic antimicrobial peptides and increases resistance to outer membrane perturbation. Antimicrobial Agents Chemother. https://doi.org/10.1128/AAC.02590-14
Shen B, Song J, Zhao Y, Zhang Y, Liu G, Li X, Guo X, Li W, Cao Z, Wu Y (2018) Triintsin, a human pathogenic fungus-derived defensin with broad-spectrum antimicrobial activity. Peptides. https://doi.org/10.1016/j.peptides.2018.08.003
Shin JM, Gwak JW, Kamarajan P, Fenno JC, Rickard AH, Kapila YL (2016) Biomedical applications of nisin. J Appl Microbiol. https://doi.org/10.1111/jam.13033
Simons A, Alhanout K, Duval RE (2020) Bacteriocins, antimicrobial peptides from bacterial origin: overview of their biology and their impact against multidrug-resistant bacteria. Microorganisms. https://doi.org/10.3390/microorganisms8050639
Singh A, Duche RT, Wandhare AG, Sian JK, Singh BP, Sihag MK, Singh KS, Sangwan V, Talan S, Panwar H (2023) Milk-derived antimicrobial peptides: overview, applications, and future perspectives. Probiotics Antimicrobial Prot. https://doi.org/10.1007/s12602-022-10004-y
Solanki SS, Singh P, Kashyap P, Sansi MS, Ali SA (2021) Promising role of defensins peptides as therapeutics to combat against viral infection. Microbial Pathog. https://doi.org/10.1016/j.micpath.2021.104930
Soleymani-Goloujeh M, Nokhodchi A, Niazi M, Najafi-Hajivar S, Shahbazi-Mojarrad J, Zarghami N, Zakeri-Milani P, Mohammadi A, Karimi M, Valizadeh H (2018) Effects of N-terminal and C-terminal modification on cytotoxicity and cellular uptake of amphiphilic cell penetrating peptides. Artif Cells Nanomed Biotechnol. https://doi.org/10.1080/21691401.2017.1414823
Di Somma A, Canè C, Moretta A, Duilio A (2021) Interaction of temporin-l analogues with the E.Coli ftsz protein. Antibiotics. https://doi.org/10.3390/antibiotics10060704
Szymczak P, Możejko M, Grzegorzek T, Jurczak R, Bauer M, Neubauer D, Sikora K, Michalski M, Sroka J, Setny P, Kamysz W, Szczurek E (2023) Discovering highly potent antimicrobial peptides with deep generative model HydrAMP. Nat Commun. https://doi.org/10.1038/s41467-023-36994-z
Tajbakhsh M, Karimi A, Fallah F, Akhavan MM (2017) Overview of ribosomal and non-ribosomal antimicrobial peptides produced by Gram positive bacteria. Cell Mol Biol. https://doi.org/10.14715/cmb/2017.63.10.4
Talat A, Khan AU (2023) Artificial intelligence as a smart approach to develop antimicrobial drug molecules: a paradigm to combat drug-resistant infections. Drug Discov Today 28(4):103491. https://doi.org/10.1016/j.drudis.2023.103491
Tall YA, Al-Rawashdeh B, Abualhaijaa A, Almaaytah A, Masadeh M, Alzoubi KH (2020) Functional characterization of a novel hybrid peptide with high potency against gram-negative bacteria. Curr Pharm Des 26(3):376–385. https://doi.org/10.2174/1381612826666200128090700
Tam JP, Wang S, Wong KH, Tan WL (2015) Antimicrobial peptides from plants. Pharmaceuticals. https://doi.org/10.3390/ph8040711
Tang C, Miller AF, Saiani A (2014) Peptide hydrogels as mucoadhesives for local drug delivery. Int J Pharm. https://doi.org/10.1016/j.ijpharm.2014.02.039
Tanhaieian A, Sekhavati MH, Ahmadi FS, Mamarabadi M (2018) Heterologous expression of a broad-spectrum chimeric antimicrobial peptide in Lactococcus lactis: its safety and molecular modeling evaluation. Microbial Pathog. https://doi.org/10.1016/j.micpath.2018.09.016
Ting DSJ, Beuerman RW, Dua HS, Lakshminarayanan R, Mohammed I (2020) Strategies in translating the therapeutic potentials of host defense peptides. Front Immunol. https://doi.org/10.3389/fimmu.2020.00983
de la Torre C, Agostini A, Mondragón L, Orzáez M, Sancenón F, Martínez-Máñez R, Marcos MD, Amorós P, Pérez-Payá E (2014) Temperature-controlled release by changes in the secondary structure of peptides anchored onto mesoporous silica supports. Chem Commun 50(24):3184–3186. https://doi.org/10.1039/C3CC49421G
Trimble MJ, Mlynárčik P, Kolář M, Hancock REW (2016) Polymyxin: alternative mechanisms of action and resistance. Cold Spring Harbor Perspect Med. https://doi.org/10.1101/cshperspect.a025288
Tripathi S, Wang G, White M, Qi L, Taubenberger J, Hartshorn KL (2015) Antiviral activity of the human cathelicidin, LL-37, and derived peptides on seasonal and pandemic influenza a viruses. PLoS ONE. https://doi.org/10.1371/journal.pone.0124706
Vanzolini T, Bruschi M, Rinaldi AC, Magnani M, Fraternale A (2022a) Multitalented synthetic antimicrobial peptides and their antibacterial, antifungal and antiviral mechanisms. Int J Mol Sci 23(1):545. https://doi.org/10.3390/ijms23010545
Vanzolini T, Bruschi M, Rinaldi AC, Magnani M, Fraternale A (2022b) Multitalented synthetic antimicrobial peptides and their antibacterial, antifungal and antiviral mechanisms. Int J Mol Sci. https://doi.org/10.3390/ijms23010545
Velkov T, Thompson PE, Azad MAK, Roberts KD, Bergen PJ (2019) History, Chemistry and antibacterial spectrum. Adv Exp Med Biol. https://doi.org/10.1007/978-3-030-16373-0_3
Vilas Boas LCP, Campos ML, Berlanda RLA, de Carvalho Neves N, Franco OL (2019) Antiviral peptides as promising therapeutic drugs. Cell Mol Life Sci. https://doi.org/10.1007/s00018-019-03138-w
Wade HM, Darling LE, Elmore DE (2019) Hybrids made from antimicrobial peptides with different mechanisms of action show enhanced membrane permeabilization. Biochimica Et Biophysica Acta (BBA)—Biomembr. https://doi.org/10.1016/j.bbamem.2019.05.002
Walter A, Mayer C (2019) Peptidoglycan structure, biosynthesis, and dynamics during bacterial. Growth. https://doi.org/10.1007/978-3-030-12919-4_6
Wang S, Zeng X, Yang Q, Qiao S (2016) Antimicrobial peptides as potential alternatives to antibiotics in food animal industry. Int J of Mol Sci. https://doi.org/10.3390/ijms17050603
Wang C, Yang C, Chen Y, Ma L, Huang K (2019) Rational design of hybrid peptides: a novel drug design approach. Curr Med Sci 39(3):349–355. https://doi.org/10.1007/s11596-019-2042-2
Wang Y, Chang RYK, Britton WJ, Chan HK (2022) Advances in the development of antimicrobial peptides and proteins for inhaled therapy. Adv Drug Deliv Rev. https://doi.org/10.1016/j.addr.2021.114066
Wang L, Qin T, Zhang Y, Zhang H, Hu J, Cheng L, Xia X (2023) Antimicrobial peptides from fish: main forces for reducing and substituting antibiotics. Turkish J Fisheries Aquatic Sci. https://doi.org/10.4194/TRJFAS23922
Wątły J, Miller A, Kozłowski H, Rowińska-Żyrek M (2021) Peptidomimetics—an infinite reservoir of metal binding motifs in metabolically stable and biologically active molecules. J Inorganic Biochem. https://doi.org/10.1016/j.jinorgbio.2021.111386
Wu W, Wang W, Li J (2015) Star polymers: advances in biomedical applications. Progress Polym Sci. https://doi.org/10.1016/j.progpolymsci.2015.02.002
Xu X, Lai R (2015) The chemistry and biological activities of peptides from amphibian skin secretions. Chem Rev 115(4):1760–1846. https://doi.org/10.1021/cr4006704
Xu D, Lu W (2020) Defensins: a double-edged sword in host immunity. Front Immunol. https://doi.org/10.3389/fimmu.2020.00764
Xuan J, Feng W, Wang J, Wang R, Zhang B, Bo L, Chen ZS, Yang H, Sun L (2023) Antimicrobial peptides for combating drug-resistant bacterial infections. Drug Resist Updates. https://doi.org/10.1016/j.drup.2023.100954
Yang M, Liu S, Zhang C (2023) Antimicrobial peptides with antiviral and anticancer properties and their modification and nanodelivery systems. Curr Res Biotechnol. https://doi.org/10.1016/j.crbiot.2023.100121
Yasir M, Dutta D, Hossain KR, Chen R, Ho KKK, Kuppusamy R, Clarke RJ, Kumar N, Willcox MDP (2020) Mechanism of action of surface immobilized antimicrobial peptides against Pseudomonas aeruginosa. Front Microbiol. https://doi.org/10.3389/fmicb.2019.03053
Yeo J, Peeva L, Chung S, Gaffney P, Kim D, Luciani C, Tsukanov S, Seibert K, Kopach M, Albericio F, Livingston A (2021) Liquid phase peptide synthesis via one-pot nanostar sieving (PEPSTAR). Angew Chem—Int Ed. https://doi.org/10.1002/anie.202014445
Yi HY, Chowdhury M, Huang YD, Yu XQ (2014) Insect antimicrobial peptides and their applications. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-014-5792-6
Yu G, Baeder DY, Regoes RR, Rolff J (2018) Predicting drug resistance evolution: insights from antimicrobial peptides and antibiotics. Proceed Royal Soc b: Biol Sci. https://doi.org/10.1098/rspb.2017.2687
Zhang C, Yang M (2022) Antimicrobial peptides: from design to clinical application. Antibiotics 11(3):349. https://doi.org/10.3390/antibiotics11030349
Zhang RY, Thapa P, Espiritu MJ, Menon V, Bingham JP (2018) From nature to creation: going around in circles, the art of peptide cyclization. Bioorganic Med Chem. https://doi.org/10.1016/j.bmc.2017.11.017
Zhang X, Zhao L, Zhai G, Ji J, Liu A (2019) Multifunctional polyethylene glycol (PEG)-Poly (Lactic-Co-Glycolic Acid) (PLGA)-Based nanoparticles loading doxorubicin and tetrahydrocurcumin for combined chemoradiotherapy of glioma. Med Sci Monit 25:9737–9751. https://doi.org/10.12659/MSM.918899
Zhang QY, Yan ZB, Meng YM, Hong XY, Shao G, Ma JJ, Cheng XR, Liu J, Kang J, Fu CY (2021a) Antimicrobial peptides: mechanism of action, activity and clinical potential. Military Med Res. https://doi.org/10.1186/s40779-021-00343-2d
Zhang Q-Y, Yan Z-B, Meng Y-M, Hong X-Y, Shao G, Ma J-J, Cheng X-R, Liu J, Kang J, Fu C-Y (2021b) Antimicrobial peptides: mechanism of action, activity and clinical potential. Mil Med Res 8(1):48. https://doi.org/10.1186/s40779-021-00343-2
Zhang Y, Towers CG, Singh UK, Liu J, Håkansson M, Logan DT, Donini O, Kutateladze TG (2022) Dusquetide modulates innate immune response through binding to p62. Structure. https://doi.org/10.1016/j.str.2022.05.003
Zhao H, To KKW, Sze KH, Yung TTM, Bian M, Lam H, Yeung ML, Li C, Chu H, Yuen KY (2020) A broad-spectrum virus-and host-targeting peptide against respiratory viruses including influenza virus and SARS-CoV-2. Nat Commun. https://doi.org/10.1038/s41467-020-17986-9
Zhou C, Wang F, Chen H, Li M, Qiao F, Liu Z, Hou Y, Wu C, Fan Y, Liu L, Wang S, Wang Y (2016) Selective antimicrobial activities and action mechanism of micelles self-assembled by cationic oligomeric surfactants. ACS Appl Mater Interfaces. https://doi.org/10.1021/acsami.5b12688
Zhu Q, Chen Z, Paul PK, Lu Y, Wu W, Qi J (2021) Oral delivery of proteins and peptides: challenges, status quo and future perspectives. Acta Pharmaceutica Sinica B 11(8):2416–2448. https://doi.org/10.1016/j.apsb.2021.04.001
Funding
No funding has been received to any author of this paper.
Author information
Authors and Affiliations
Contributions
A . Vrushali Somase- As the main author of this review article I made substantial contributions to various aspects of the manuscript. I have written the whole manuscript including Conceptualization, Literature Review, and Draft writing. B Dr. Sharav Desai- Guidance C Dr. Vipul Patel- guidance D Vivek Patil- prepared Table E Kunal Bhosale- Prepared Figures
Corresponding author
Ethics declarations
Competing Interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Somase, V., Desai, S.A., Patel, V.P. et al. Antimicrobial Peptides: Potential Alternative to Antibiotics and Overcoming Limitations for Future Therapeutic Applications. Int J Pept Res Ther 30, 45 (2024). https://doi.org/10.1007/s10989-024-10623-9
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-024-10623-9