
Abstract
In this paper, the crystallization kinetics of Hf26Be18Ti18Zr18Cu7.5Ni12.5 high mixing entropy amorphous alloys under non-isothermal conditions are studied. The alloy shows two different crystallization events. In addition, the activation energies of the two crystallization events are calculated using the Kissinger, Augis-Bennett, and Ozawa methodologies. Similar values are obtained by the three equations. The activation energy of the first crystallization event is slightly less than that of the second crystallization events, which indicates that the first crystallization can easily occur. The modified Johnson–Mehl–Avrami (JMA) equation is then used to further analyze the non-isothermal crystallization kinetics. The Avrami exponent (n(α)) is between 1.5 and 2.5 for the first crystallization even and most instances (0.1 < α < 0.5) of the second crystallization event, which demonstrates that the crystallization mechanism has mainly been controlled by a three-dimensional growth with a nucleation rate decrease. Moreover, n(α) is between 1 and 1.5 in the second stage of the second crystallization event (0.5 < α < 0.9), which implies a direct growth of crystal nuclei. Compared with the other alloys, Hf26Be18Ti18Zr18Cu7.5Ni12.5 has stronger high entropy effect, leading to more sluggish diffusion and more difficult crystallization.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Due to the lack of dislocations, grain boundaries, and other defects in the amorphous alloy, it has high strength, high hardness, high wear resistance, and other unique properties, that currently attract more attention [1,2,3,4]. The previously developed amorphous alloys are multicomponent alloys containing one or two principal elements [5,6,7,8]. In recent years, with the development of the amorphous alloy concept, several kinds of equiatomic or near equiatomic amorphous alloys that are defined as high entropy amorphous alloys, have been presented [9]. High entropy amorphous alloys have high glass-forming ability (GFA) and unique mechanical properties compared with conventional amorphous alloys. This provides a new idea for the discovery of new amorphous alloy systems and is of great significance for their application. For example, the Pd20Pt20Cu20Ni20P20 high entropy amorphous alloys [9] have a large GFA with a diameter of 10 mm. Zhao [10] recently studied the mechanical properties and the glass-forming ability of the Ti20Zr20Hf20Be20(Cu20-xNix) high entropy amorphous alloys. Among those amorphous alloys, the Ti20Zr20Hf20Be20(Cu50Ni50)20 alloy has the best GFA, a critical diameter greater than 25 mm, and a yield strength up to 2019 MPa. In addition, the Hf-based high entropy amorphous alloy has larger GFA than the other alloys, its critical diameter is 15 mm, and its strength at room temperature reaches 2539 MPa [11].
According to the studies presented in [12, 13], the amorphous alloys have very good plastic deformation in the supercooled liquid region, which resolves the issue of amorphous alloys being difficult to be mechanically processed due to their low plasticity at room temperature. Amorphous alloys are generally metastable materials, and they will crystallize during high-temperature deformation, which will affect their performance. Therefore, it is very important to analyze the crystallization kinetics of amorphous alloys, which will help to understand their crystallization during high-temperature deformation. Different models are used to describe the crystallization kinetics according to the heating process. Activation energies are calculated using the Kissinger, Ozawa, and Augis-Bennett methods in non-isothermal mode [14]. The crystallization kinetics is usually studied using the Johnson–Mehl–Avrami (JMA) method [15, 16]. Several studies on the thermal properties and behaviors of amorphous alloy crystallization have been performed. However, few studies focused on the crystallization kinetics of the Hf-based high-entropy amorphous alloys.
In this paper, the crystallization behaviour of the Hf26Be18Ti18Zr18Cu7.5Ni12.5 high mixing entropy amorphous alloy in non-isothermal condition was studied. The activation energy and kinetic parameters of crystallization were calculated to explain the nucleation and growth mechanism.
Experimental
Master alloy ingots with nominal composition of Hf26Be18Ti18Zr18Cu7.5Ni12.5 (at.%) were prepared by arc melting the mixture of Hf, Be, Ti, Zr, Cu, and Ni metals (purity ≥ 99.9%) in a high-purity argon atmosphere. They were remelted 4 times to ensure the complete melting and the composition homogeneity. The cylindrical rods, having a diameter of 4 mm, were then produced by copper mold suction casting. The microstructure of Hf26Be18Ti18Zr18Cu7.5Ni12.5 was studied by X-ray diffraction (XRD, bruker D2 PHASER, Cu (Ka)) and transmission electron microscopy (TEM, JEOL JEM-2010). The non-isothermal crystallization behavior was determined using a differential scanning calorimeter (DSC, METTLER-TOLEDO TGA/DSC1) under heating rates of 10, 20, 30, 40, and 50 K min−1.
Results and discussion
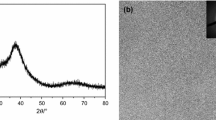
Figure 1A shows the XRD pattern of the as-cast Hf26Be18Ti18Zr18Cu7.5Ni12.5 amorphous alloy. A broad diffuse scattering peak in the range of 35–50° can be observed, indicating that the alloy has a fully amorphous structure. Figure 1b shows TEM micrographs of the Hf26Be18Ti18Zr18Cu7.5Ni12.5 alloy. A uniform microstructure can be observed from the bright-field image. In addition, the consequent selected area of the electron diffraction pattern exhibits an amorphous ring, which shows a fully amorphous structure.

XRD pattern (a) and TEM images (b) of the as-cast Hf26Be18Ti18Zr18Cu7.5Ni12.5
The DSC curves of Hf26Be18Ti18Zr18Cu7.5Ni12.5 obtained at different heating rates are shown in Fig. 2. The representative temperatures, including the glass transition (Tg), onset crystallization (Tx), and crystallization peak (Tp) temperatures, are indicated by arrows. The DSC curves have two exothermic peaks that are related to the crystallization of the Hf26Be18Ti18Zr18Cu7.5Ni12.5 amorphous alloy. The characteristic temperatures, such as Tg, Tx1, Tp1, Tx2, and Tp2, as well as the temperature difference △Tx (△Tx = Tx–Tg), are presented in Table 1. The results show that these characteristic temperatures increase with the increase of the heating rate, which indicates that there is a significant correlation between the crystallization rate and the heating rate.

DSC curves of Hf26Be18Ti18Zr18Cu7.5Ni12.5 at different heating rates
E may be used to represent the difficulty of crystallization, which can be calculated using the Kissinger, Augis–Bennett, and Ozawa equations. The Kissinger equation is given by [17]:
where β denotes the heating rate, R represents the constant, and T is the temperature.
E can also be calculated using the Ozawa equation [18]:
Finally, the Augis–Bennett equation is given by [19]:
It can be observed that Eq. (3) describes a linear relationship between 1/T and ln(T/β).
Equations (1), (2), and (3) are then used to calculate the activation energies of the Hf26Be18Ti18Zr18Cu7.5Ni12.5 amorphous alloy. The Kissinger, Ozawa, and Augis–Bennett plots are shown in Fig. 3. Ex1, Ep1, Ex2, and Ep2 are shown in Table 2. It can be observed that the activation energies calculated using the three equations are very similar. However, the activation energies calculated using Eq. (2) are larger than those calculated using Eqs. (1) and (3). Hf26Be18Ti18Zr18Cu7.5Ni12.5 has larger Ep and Ex values than Hf20Ti20Zr20Be20(Cu10Ni10) (Ex1 = 262 kJ mol−1, Ep1 = 242.6 kJ mol−1, Ex2 = 492.8 kJ mol−1, and Ep2 = 483 kJ mol−1) [20], which demonstrates that it has a high thermodynamic stability.

Kissinger (a), Ozawa (b), and Augis–Bennett (c) plots for Hf26Be18Ti18Zr18Cu7.5Ni12.5
Tx is related to the nucleation process. In addition, it can be observed that Tp is also related to its growth process [21]. Therefore, Ex1 and Ex2 can represent the activation energy of grain nucleation, while Ep1 and Ep2 can represent the activation energy of the growth process. It can be deduced from Table 2 that Ex1 is lower than Ex2, which implies that the barrier of energy for the second crystallization is larger than that of the first crystallization. This also indicates that the first crystallization is more likely to occur. Moreover, Ep1 is lower than Ep2, which indicates that the growth process during the first crystallization event is more likely to germinate than the second crystallization event. Finally, it can be deduced that Ep is lower than Ex, which indicates that the nucleation is more likely to occur than the grain growth during crystallization.
Using the previously described methods, it can be deduced that E is related to the first and second crystallizations. However, due to the difference of the grain growth and grain nucleation, the activation energy for distinct crystallized volume fractions (α) may change. Consequently, the dependence of the activation energy on α should be further discussed during the crystallization process.
α can be computed as [22]:
where T0 and \({T}_{\infty }\) are the temperatures of crystallization start and crystallization end in the amorphous alloys, respectively. Figure 4 presents α for two exothermic peaks for Hf26Be18Ti18Zr18Cu7.5Ni12.5. A temperature sigmoid dependency at various heating rates is observed for all the amorphous alloys during the non-isothermal crystallization processes [23], which indicates that the rate of crystallization is slower at α < 0.1 and α > 0.9. It can also be observed that the crystallization reaction quickly occurs for 0.1 < α < 0.9.

Curves of the crystallization volume fraction (α) of Hf26Be18Ti18Zr18Cu7.5Ni12.5 at different heating rates: first crystallization (a), and second crystallization (b)
The activation energies Ea(α) at different crystallized volume fractions are calculated using the Kissinger–Akahira–Sunose (KAS) equation [16, 24, 25]:
Figure 5 shows the KAS curves for the two crystallization peaks for the Hf26Be18Ti18Zr18Cu7.5Ni12.5 BMG. It is determined for the evolution of Ea(α) function of α through linear fitting (Fig. 6). It can be deduced that the average activation energies for the first and second crystallization events are 204.8 kJ.mol−1 and 222.5 kJ.mol−1, respectively. In particular, Ea(α) of the second crystallization peak is smaller than that of the first crystallization peak. Note that these results are consistent with the calculated results. The higher the Ea(α) values of the first crystallization stage, the more difficult the crystallization, which results in a slow increase of α (Fig. 4). Ea(α) starts to first increase, and it finally decreases after the beginning of the crystallization.

KAS curves for the crystallization volume fraction (α) for Hf26Be18Ti18Zr18Cu7.5Ni12.5: first crystallization (a), and second crystallization (b)

Activation energy (E(α)) function of the crystallization volume fraction (α) in non-isothermal conditions
The isothermal crystallization kinetics can be derived from the JMA equation [26]:
where \(\alpha\) is the crystallized volume fraction, t represents the annealing time, and \(\tau\) denotes the incubation time. However, the JMA equation can only be used for isothermal modes. Blázquez [27] proposed an extended version of the JMA equation for non-isothermal conditions:
where \({T}_{0}\) represents the initial temperature crystallization and \({E}_{\text{a}}\) is the corresponding activation energy.
Figure 7 shows the curves of \(\mathrm{ln}\left([-\mathrm{ln}(1-\alpha )]\right)\) and \([\mathrm{ln}\left(T-{T}_{0}\right)/\beta ]\) for Hf26Be18Ti18Zr18Cu7.5Ni12.5. Based on Eq. (7) and Fig. 7, n(α) varies with respect to the crystallized fraction (α), as shown in Fig. 8. For the first crystallization event, the n value reaches 2.5 at α = 0.1 at the beginning of the crystallization process. When α increases, n decreases. At α = 0.9, n is mostly larger than 1.5. It can be observed that in almost the entire crystallization process, n(α) ranges between 1.5 and 2.5 for 0.1 < α < 0.9, which indicates that the crystallization process is dominated by diffusion-controlled three-dimensional growth with the decrease of the nucleation rate [13,14,15]. For the second crystallization event, n(α) ranges between 1.5 and 2.5. However, the second crystallization should have two steps. When α is in the range of 0.1–0.5, n ranges between 1.5 and 2, which indicates that the crystallization mechanism is guided to diffusion-controlled three-dimensional growth with the decrease of the nucleation rate. When α is in the range of 0.5–0.9, n is less than 1.5, which indicates that a more crystal nuclei directly grows up [13,14,15]. The second crystallization of the amorphous alloy can be divided into two steps: precipitation of the small crystallites, and growth and aggregation of the crystal grains.

\(\mathrm{ln}\left([-\mathrm{ln}(1-\alpha )]\right)\) versus \([\mathrm{ln}\left(T-{T}_{0}\right)/\beta ]\) for Hf26Be18Ti18Zr18Cu7.5Ni12.5: first crystallization (a), and second crystallization (b)

Relationship between n(α) and α at different heating rates of Hf26Be18Ti18Zr18Cu7.5Ni12.5: first crystallization (a), and second crystallization (b)
In order to analyze the phase precipitation during the two crystallization events for Hf26Be18Ti18Zr18Cu7.5Ni12.5, the samples are annealed at 720 K and 784 K for 10 min. The XRD patterns of the annealed sample are shown in Fig. 9. The alloy annealed at 720 K (Tp1) generates a small number of diffraction peaks, representing the precipitation of FCC and BCC solid solutions. When the annealing temperature increases to 784 K, the Ni7Zr2 and Cu5Zr are also detected in the XRD curves, in addition to BCC and FCC solid solutions. Figure 9 shows that completely crystallized Hf26Be18Ti18Zr18Cu7.5Ni12.5 samples are detected in the four crystalline phases. However, HfTiZrBeCu and HfTiZrBeNi high-entropy amorphous alloys [27] can be detected in only three phases. Hf26Be18Ti18Zr18Cu7.5Ni12.5 has stronger high entropy effect, leading to more sluggish diffusion and more difficult crystallization.

XRD patterns of the annealed Hf26Be18Ti18Zr18Cu7.5Ni12.5
Conclusions
In this paper, the non-isothermal crystallization kinetics of Hf26Be18Ti18Zr18Cu7.5Ni12.5 high mixing entropy amorphous alloys were investigated by XRD and DSC. The conclusions are as follows:
-
1.
Hf26Be18Ti18Zr18Cu7.5Ni12.5 had two distinct crystallization events. In addition, the characteristic temperatures (Tx1, Tp1, Tx2, and Tp2) increased with the increase of the heating rate.
-
2.
The activation energy was calculated using the Kissinger, Augis–Bennett, and Ozawa methods, and similar values were obtained. It was observed that the activation energy of the first crystallization event is slightly lower than that of the second crystallization event. This indicates that the first crystallization can easily occur.
-
3.
The local Avrami exponent value (n(α)) ranges between 1.5 and 2.5 for the first crystallization even and most of the instances (0.1 < α < 0.5) of the second crystallization event, which indicates that the crystallization mechanism is guided to diffusion-controlled three-dimensional growth with the decrease of the nucleation rate. In the second stage (0.5 < α < 0.9), for the second crystallization event, n(α) ranges between 1 and 1.5, which indicates that a more crystal nuclei directly grows up.
-
4.
Hf26Be18Ti18Zr18Cu7.5Ni12.5 shows four crystalline phases in the corresponding complete crystallization. The alloy demonstrates stronger high entropy effect, leading to more sluggish diffusion and more difficult crystallization.
-
5.
Due to the fact that the sluggish diffusion remains amorphous during heat processes at a temperature less than Tp1, Hf26Be18Ti18Zr18Cu7.5Ni12.5 can be used for various applications such as the precise instrument and biomedical field in different crystallization temperature ranges.
References
Inoue A, Wang XM, Zhang W. Developments and applications of bulk metallic glasses. Rev Adv Mater Sci. 2008;18:1–9.
Inoue A. High strength bulk amorphous alloys with low critical cooling rates(Overview). Mater Trans JIM. 1995;36(7):866–75.
Inoue A. Stabilization of metallic supercooled liquid and bulk amorphous alloys. Acta Mater. 2000;48:279–306.
Inoue A, Zhang T, Saida J, Matsushita M, Chen MW, Sakurai T. High strength and good ductility of bulk quasicrystalline base alloys in Zr65Al7.5Ni10Cu7.5-xPdx system. Mater Trans JIM. 1999;40(10):1137–43.
Shcheretskyy OA, Lakhnenko VL, Shumikhin VS, Bespalyy AA, Soloviova AV. Fabrication of nanostructural materials by means of heattreatment of amorphous Zr64Cu16Ni10Al9.5Nb0.5 alloy Metallofiz. Noveishie Tekhnol. 2011;33(10):1323–32.
Pelletier JM, Louzguine-Luzgin DV, Li S, Inoue A. Elastic and viscoelastic properties of glassy quasicrystalline and crystalline phases in Zr65Cu5Ni10Al7.5Pd12.5 alloys. Acta Mater. 2011;59:2797–806.
Sun BR, Xin SW, Shen TD. Low-temperature magnetization and magnetic exchange interactions in Fe40Ni40P14B6 bulk metallic glasses. J Magn Magn Mater. 2017;429:276–80.
Aronhime N, DeGeorge V, Keylin V, Ohodnicki P, McHenry ME. The effects of strain-annealing on tuning permeability and lowering losses in Fe-Ni-based metal amorphous nanocomposites. JOM. 2017;69:2164–70.
Takeuchi A, Chen N, Wada T, Yokoyama Y, Kato H, Inoue A. Pd20Pt20Cu20Ni20P20 high-entropy alloy as a bulk metallic glass in the centimeter. Intermetallics. 2011;19:1546–54.
Gong P, Yao KF, Ding HY. Crystallization kinetics of HfTiZrCuNiBe high entropy bulk metallic glass. Mater Lett. 2015;156:146–9.
Zhao SF, Shao Y, Liu X, Chen N, Ding HY, Yao KF. Pseudoquinary Ti20Zr20Hf20Be20(Cu20-xNix) high entropy bulk metallic glasses with large glass forming ability. Mater Des. 2015;87:625–31.
Cao QP, Liu JW, Li JF, Zhou YH, Wang XD, Jiang JZ. Isochronal crystallization kinetics of Cu60Zr20Ti20 bulk metallic glass. J Non Cryst Solids. 2011;357:1182–7.
Li B, Li Yh, Yang K, Li JS, Fan XH. Effect of yttrium addition on the non-isothermal crystallization kinetics and fragility of Cu– Zr–Al bulk metallic glass. Thermochim Acta. 2016;642:105–10.
Wang XF, Wang D, Zhu B, Li YJ, Han FS. Crystallization kinetics and thermal stability of mechanically alloyed Al76Ni8Ti8Zr4Y4 glassy powder. J Non Cryst Solids. 2014;385:111–6.
Cui J, Li JS, Wang J, Kou HC, Qiao JC, Gravierc S, Blandinc JJ. Crystallization kinetics of Cu38Zr46Ag8Al8 bulk metallic glass in different heating conditions. J Non Cryst Solids. 2014;404:7–12.
Hu XX, Jichao Q, Pelletier JM, Yao Y. Evaluation of thermal stability and isochronal crystallization kinetics in the Ti40Zr25Ni8Cu9Be18 bulk metallic glass. J Non Cryst Solids. 2016;432:254–64.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29:1702–6.
Ozawa T. Kinetic analysis of derivative curves in thermal analysis. J Therm Anal Calorim. 1970;2:301–24.
Augis JA, Bennett JE. Calculation of the Avrami parameters for heterogeneous solid state reactions using a modifification of the Kissinger method. J Therm Anal Calorim. 1978;13:283–99.
Gong P, Li FW, Yin G, Deng L, Wang XY, Jin JS. Thermal cycling effect on the kinetics of glass transition and crystallization of a Zr-based bulk metallic glass. J Therm Anal Calorim. 2020;142:63–73.
Zhuang YX, Duan TF, Shi HY. Calorimetric study of nonisothermal crystallization kinetics of Zr60Cu20Al10Ni10 bulk metallic glass. J Alloys Compd. 2011;509:9019–25.
Zhang LC, Xu J, Eckert J. Thermal stability and crystallization kinetics of mechanically alloyed TiC/Ti-based metallic glass matrix composite. J Appl Phys. 2006;100:033514.
Cao QP, Li JW, Li JF, Zhou YH, Wang XD, Jiang JZ. Isochronal crystallization kinetics of Cu60Zr20Ti20 bulk metallic glass. J NonCryst Solids. 2011;357:1182–7.
Prajapati SR, Sauthor K, Ashmi TP, Pratap A. Non-isothermal crystallization kinetics of Zr52Cu18Ni14Al10Ti6 metallic glass. J Therm Anal Calorim. 2016;124:21–33.
Ranganathan S, Heimendahl MV. The three activation energies with isothermal transformations: applications to metallic glasses. J Mater Sci. 1981;16:2401–4.
Wang WH, Dong C, Shek CH. Bulk metallic glasses. Mater Sci Eng R Rep. 2004;44(2):45–89.
Jin JS, Li FW, Yin G, Wang XY, Gong P. Influence of substitution of Cu by Ni on the crystallization kinetics of TiZrHfBeCu high entropy bulk metallic glass. Thermochim Acta. 2020;690:178650.
Acknowledgements
This work was financially supported by the General Research Project of Shaanxi Provincial Education Department (No. 22JK0418).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, K., Li, B., Fan, Xh. et al. Investigation of crystallization behavior of Hf26Be18Ti18Zr18Cu7.5Ni12.5 high mixing entropy amorphous alloys. J Therm Anal Calorim 148, 689–696 (2023). https://doi.org/10.1007/s10973-022-11778-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-022-11778-7