
Abstract
A model is proposed to fit differential scanning calorimetry (DSC) isothermal crystallization curves obtained from the molten state at different temperatures. A commercial 3D printing polylactic acid (PLA) sample is used to test the method. All DSC curves are fitted by a mixture of two simultaneous functions, one of them being a time derivative generalized logistic accounting for the exothermic effect and the other, a generalized logistic, accounting for the baseline. There is a rate parameter, which is allowed to vary across different temperatures. The rate parameter values obtained at different temperatures were jointly explained as a result of three crystallization processes, each one defined by a characteristic crystallization time, a characteristic temperature, and a dispersion or width factor. Apart from the very good fittings obtained at all temperatures, the results agree with the existence of a few crystal forms of PLA, which were demonstrated by other authors. Thus, the main significance of this work consists in providing a new approach in order to mathematically describe the isothermal crystallization kinetics of a polymer from the melt. Such a kinetic description is needed in order to predict the extent of a crystallization process as a function of time at any isothermal temperature. The approach used here allows to understand the overall crystallization of the PLA used in this work as the sum of three crystallization processes, each of them corresponding to a different crystal form. Each experimental crystallization exotherm, which may include more than one crystal form, can be reproduced by a generalized logistic function. The overall rate factor at a given temperature is the weighted sum of the rate factors of the different crystal structures at that temperature. The rate factor of each of these three processes is described by a Gaussian function whose parameters are a crystallization time, a characteristic temperature and a temperature dispersion factor. Therefore, the crystallization rate for each crystal form can be interpreted as a relative likelihood to crystallize at a given temperature. On the other hand, the characteristic crystallization time parameter refers to the time needed for a given crystal structure to be formed at the temperature at which the relative likelihood to crystallize of that form is highest.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Crystallization of polymers from the melt is usually complex because there are more than one possible crystalline structure. A typical example of complex crystallization is that of polylactic acid (PLA) [1,2,3]. Because of this, the crystallization peaks may have shoulders depending on the cooling conditions, as those reported by some of the authors in previous works [4, 5]. Model-fitting and model-free kinetic methods have been largely discussed and become classical approaches [6,7,8,9]. A model-fitting approach assumes a fixed mechanism throughout the reaction, while model-free methods allow to evaluate Arrhenius parameters without choosing the reaction model [9]. The best known representatives of the model-free approach are the isoconversional methods. According to the isoconversional principle, the reaction rate at constant extent of conversion is only a function of temperature. These methods yield the effective activation energy as a function of the extent of conversion. Both differential and integral isoconversional methods have been developed. A common differential isoconversional method is that of Friedman [10]. It was reported that when working with differential data obtained from DSC, some inaccuracy may arise as it is difficult to determine the correct baseline [11]. Also, incremental isoconversional methods have been developed [12, 13]. According to some reports, the activation parameters represent apparent quantities without a mechanistic interpretation [13]. The transition zone theory accurately describes crystal growth rates from the glass transition temperature to the melting temperature [14].
In spite of all the very good approaches to evaluate kinetics from thermoanalytical data, the authors are not aware of a model that allows to accurately fit a set of isothermal DSC crystallization curves corresponding to a broad range of temperatures at all temperatures in practically all the extension of the isothermal data. Testing of multiple samples is generally recommended to extract a crystal growth rate constant, assuming that a single crystal form is observed [15,16,17]. However, in this study, it is not assumed that a single-crystal form has to exist. No attempt is made to separate nucleation from growth. Instead, it is considered as a global crystallization process that encompasses the possible processes of nucleation and growth of the different crystalline forms that may arise in an industrial polymer such as the one used in this work. For the present study, it is critical to minimize the experimental factors to see better relations between the measured signal and the model parameters. This work is focused on the study of isothermal crystallization of polylactic acid (PLA) from the molten state, covering a broad range of temperatures between its glass transition temperature, Tg, and its melting temperature, Tm. The approach used here consists of fitting each exotherm by a mixture of two simultaneous functions, one accounting for the exothermic effect and the other for the baseline. Then the rate parameter values obtained at each temperature are jointly analyzed and explained as a result of multiple crystallization processes, each one defined by a characteristic crystallization time, a characteristic temperature, and a dispersion or width factor.
Experimental
The material used for all samples was a commercial PLA from Ultimaker. This PLA was supplied in the form of filament, with a diameter of 2.85 mm. 3DP filaments may include nucleating agents and other additives. In this particular case, the composition was not disclosed.
A preliminary thermogravimetric (TG) analysis of the sample was performed on a TA Instruments 2960 SDT. The experiment consisted of a linear heating ramp at 20 ºC min−1 from room temperature to 1200 ºC. A 100 mL min−1 dry air purge was kept along the experiment.
DSC tests were performed with a single sample on a TA Instruments Q2000 MDSC. The experiments consisted of a linear heating at 30 ºC min−1 up to 170 ºC followed by a 2-min isothermal step and then a cooling ramp at 30 ºC min−1 down to the crystallization temperature. Then, the temperature remained constant until a flat baseline was obtained after the exotherm. This cycle was repeated for 30 crystallization temperatures, some of which were randomly repeated to check the repeatability of the results.
Results
The result of the preliminary TG test is shown in Fig. 1. It is similar to reported results from neat PLA, with the exception that here there is a small loss of mass in the range from 400 to 500 °C [18]. This small mass loss can be related to the presence of nucleating agents.

TG plot obtained from the PLA under study in air
DSC crystallization results consist of typical exotherms and are presented along with their fittings in “Appendix”. It is observed that the peak area decreases at the lower crystallization temperatures.
Figure 2 plots the heat flow curves obtained on heating subsequent to isothermal crystallization at the indicated temperatures. It can be observed how the melting peak changes as a function of the temperature of the isothermal step. It is noticeable that in the case of the ramps following the lowest isothermal temperatures a clear shoulder appears, which is consistent with the existence of more than one crystalline form of PLA. Figure 3 plots the temperatures at which the melting peaks and the shoulders are observed versus the temperature at which the sample was previously crystallized. It is observed that both the melting temperature and the shoulder depend on the crystallization temperature in a slightly different way than that reported for syndiotactic polypropylene [19]. A continual shift of the melting peak temperature subsequent to isothermal crystallization was reported for PLA [20]. Differences of about 12 ºC in the melting peak were reported by other authors [21]. In this work, a difference of 17.6 ºC was found, which is not surprising considering that the commercial PLA used here is different from that used in the reported study. In addition to the appearance of shoulders, it was also observed that the shapes of the melting endotherms were different depending on the temperature at which the crystallization was achieved. This is an indication that several crystal forms probably coexist.

Plots of the heat flow curves obtained on heating, at 30 ºC min−1, subsequent to isothermal crystallization at the indicated temperatures

Plot of the melting peak temperature, Tmeltp, and the temperature at the shoulder peak, Tshoulder, versus the temperature at which the sample was previously crystallized, Tcryst
Description of a single DSC curve
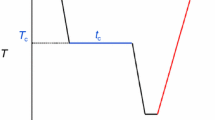
The model used to represent the differential scanning calorimetry (DSC) curves resulting from the isothermal crystallization experiments is shown in Fig. 4. A horizontal and flat baseline is expected before and after the crystallization since the temperature is constant and there is no change in the sample’s Cp. However, there is a little decrease in Cp during the crystallization associated with a decrease in the molecular mobility resulting from the crystallization process. That change in Cp is usually very small, but it may affect the fitting. In this model, it is assumed that the exothermic effect and the effect from the change of Cp are simultaneous since both result from the crystallization. The exotherm is represented by a time-derivative generalized logistic function (DGL), y2, and the baseline resulting from Cp by a generalized logistic (GL), y1. The common parameters in both functions, τ, tapm, and b, share the same values in order to ensure absolute simultaneity.
where tapm is the time at the peak maximum, c represents the area of the peak, and τ is the symmetry factor, where τ = 1 means perfect symmetry and b is a rate factor, which depends on temperature. The exotherms obtained in this work did not show any important skewness and thus τ was set to 1. The Cp change signal is obtained by the product of y1 by a scale factor, sf, that represents the total Cp change during the crystallization process. Then, each isothermal crystallization curve was fitted by a mixture of y2(t) and y1(t) where all parameters could vary except for τ, which was set to 1. The position of the curve on the y-axis is determined by the a0 parameter.
It is important to realize that data recorded prior to achieving the isothermal condition were not taken into account for the fittings.

Illustration of the Cp and exothermic contributions to the heat flow obtained in a crystallization process
DSC curve fitting
While Fig. 4 represents an ideal scenario where there is a flat baseline both before and after the transition, in practice it is not easy to determine precisely when the isothermal data following a temperature ramp section start to be valid. Figure 5 plots the heat flow and temperature curves of three experiments obtained at 80, 100, and 140 ºC. In the three cases, it can be observed how even after reaching the isothermal temperature there is some decrease in the baseline and after a little time the heat flow curve starts to rise. That kind of effect, related to the change from ramp to isothermal conditions, appears in all experiments. In general, the data preceding the exotherm were not used for the fittings. The other limit of the fitting range was set towards the end of the DSC curve where the heat flow is practically constant. The value of the DSC curve at that point is shown as a0 on Table 2 and it is related to the heat capacity of the sample for the same experimental run. However, if the sample is moved and replaced in the same position, that value may change, as it can be observed by comparing the a0 values for two different experimental runs, as indicated in the last column in the table.

Plots of the heat flow and temperature curves obtained at the indicated temperatures along with the fitting functions
Once the a0 parameter was set, the data into the selected region, between the vertical lines in Fig. 5, are fit by Eq. 3. The fitting was done with the Fityk software by minimizing the weighted sum of squared residuals, using the Nelder—Mead algorithm [22, 23].
On the other hand, it can be observed that the fitting component representing the exotherm, y2, spans to the left more than the experimental data from which it was calculated. This means that the fitting curve represents a whole exotherm including a little part on the left of the first vertical line that could not be taken into account for the fitting because the isothermal condition was not well established yet, and thus, the experimental data in that part were meaningless. This was expected since cooling of the sample and the DSC cell from melt to isothermal temperature always takes some time. It is also not surprising that this effect is more noticeable for the experiment at 100 ºC, for which a higher crystallization rate was observed.
Crystallization extent
The parameter values resulting from the fitting of all isothermal DSC curves are displayed on “Appendix” Table 2. It is observed that the peak area, c parameter, clearly decreases from 105 to 80 ºC. One could easily think of a partial crystallization during the cooling step preceding the isotherm. However, the estimated peak accounts for a possible part of the exotherm that is missing, as can be observed in Fig. 5. Several reports comment on the existence of more than one crystal form of PLA [24, 25]. It was reported that the crystallization enthalpy of PLA is lower at low temperatures because predominance of one or another coexisting phases depends on temperature. The enthalpy ratio reported for α, which is the predominant form at 150 ºC, and α’, which is the predominant form at 80 ºC was 1.68 [21]. The results presented in Table 2 are in line with that report: the ratio of the c values at 140 ºC and 80 ºC is 1.72.
The crystallization rate
The optimal fitting of 30 isothermal curves in the range of temperature between the glass transition temperature, Tg, and the melting temperature, Tm, resulted in the b values presented in Fig. 6. A trend with a double peak, similar to that of this b rate factor, was previously described for the variation of the reciprocal half-time and the Malkin’s and Avrami’s rate constants as a function of crystallization temperature [26]. The reciprocal half-time was defined as the time spent from the onset of the crystallization to the point where the crystallization is 50% complete. That double-peak trend can be fitted by a mixture of bell-shaped functions.

Plot of the b parameter values obtained at different temperatures and their fitting by a mixture of three Gaussian functions
In Supaphol’s work, the double-peak variation of the reciprocal half-time with temperature was attributed to the result of the contributions from the maximum in the crystal growth rate and from the maximum in the primary homogeneous nucleation rate. Then, considering that the overall crystallization rate parameters relate, in one way or another, to the primary homogeneous nucleation rate and the crystal growth rate, the crystallization rate parameters were fit by nonlinear multivariable regression to an expression for the overall crystallization rate data [26]. That good fitting was obtained in two separated ranges of temperature as if the crystallization rates below and above 40 ºC were independent. On the other hand, more than one possible crystalline structure for PLA was reported [1, 2]. Thus, considering that there is more than one possible crystalline structure and that the predominance of one or the other depends on temperature, the b values presented in Fig. 6 can be fitted by a mixture of a few functions, each representing a crystalline form. In practice, an optimal fitting was obtained with three Gaussian functions, which can be written as
The parameters of the function are specific for each crystallization structure and represent a characteristic crystallization time, tcryst; the temperature at which the maximum rate is obtained, Tcent, and the half width at height maximum, Thwhm, which is related to how b decreases as T diverges from Tcent. Table 1 shows the parameter values of the fitting presented in Fig. 6. It can be observed that F1 represents the main contribution in the low temperature range, while F3 is predominant at higher temperatures. F2 is much smaller than F1 and F3. It is noticeable that one of the maxima stems almost entirely from the contribution of a single function, F1, while the other maximum results from the contribution of the three functions. Thus, a maximum crystallization rate can be obtained near the central temperature of the F1 structure or at about 113 ºC, a temperature at which the three structures may simultaneously grow.
An important feature of the present model is that it allows to obtain good fittings making use of just a few parameters. The overall crystallization rate parameter was formulated as dependent on characteristic temperature parameters and characteristic crystallization times, which are specific for each crystal structure. According to Table 1, the temperature at which the crystal form corresponding to F1 can crystallize at its highest crystallization rate is about 101 ºC. The crystallization rate of this form decreases to the half of its maximum when moving 11.5 ºC up or down from the central value. The crystallization time of the F3 form is longer than that of F1, indicating a slower crystallization rate, which will be centered at a temperature about 20 ºC higher than that of F1. On the other hand, the crystal form corresponding to F2 has a much longer crystallization time and may develop to a very little extent in a narrow range around 114 ºC. Even at that temperature the crystallization rates of the other crystal forms are higher.
Conclusions
A model to fit DSC isothermal crystallization curves obtained from the molten state at different temperatures was tested with a commercial PLA sample.
The overall process is accurately represented at all temperatures by a time derivative generalized logistic. The only kinetic parameter of the logistic that changes with temperature was explained as a result of the formation of three crystal forms of PLA, whose existence was demonstrated by other authors.
The contributions to the overall crystallization rate are represented by a set of three parameters, specific for each crystal form: a characteristic crystallization time, a characteristic temperature, and a dispersion or width factor. These parameters are combined through a Gaussian function that represents the variation of the rate factor with temperature for each crystal form.
References
Gracia-Fernández CA, Gómez-Barreiro S, Álvarez-García A, López-Beceiro J, Álvarez-García B, Zaragoza-Fernández S, et al. Thermal characterization of poly-l-lactide by dielectric analysis and modulated DSC. J Therm Anal Calorim. 2014;116:1273–8.
Gracia-Fernández CA, Gómez-Barreiro S, López-Beceiro J, Naya S, Artiaga R. New approach to the double melting peak of poly(l-lactic acid) observed by DSC. J Mater Res. 2012;27:1379–82.
Wei Z, Song P, Zhou C, Chen G, Chang Y, Li J, et al. Insight into the annealing peak and microstructural changes of poly(l-lactic acid) by annealing at elevated temperatures. Polymer. 2013;54:3377–84.
López-Beceiro J, Gracia-Fernández C, Artiaga R. A kinetic model that fits nicely isothermal and non-isothermal bulk crystallizations of polymers from the melt. Eur Polym J. http://linkinghub.elsevier.com/retrieve/pii/S0014305713002140.
Supaphol P. Crystallization and melting behavior in syndiotactic polypropylene: origin of multiple melting phenomenon. J Appl Polym Sci. 2001;82:1083–97.
Janković B. The pyrolysis process of wood biomass samples under isothermal experimental conditions-energy density considerations: application of the distributed apparent activation energy model with a mixture of distribution functions. Cellulose. 2014;21:2285–314.
Janković B, Adnađević B, Jovanović J. Application of model-fitting and model-free kinetics to the study of non-isothermal dehydration of equilibrium swollen poly (acrylic acid) hydrogel: thermogravimetric analysis. Thermochim Acta. 2007;452:106–15.
Li Y, Duan L, Cheng L, Yang Y, Li Y, Cheng Y, et al. Thermal analysis and crystallization kinetics of polyurethane. J Therm Anal Calorim. 2019;135:2843–8.
Vyazovkin S. Chapter 13 isoconversional kinetics. Elsevier; 2008. p. 503–38. http://linkinghub.elsevier.com/retrieve/pii/S1573437408800167.
Friedman HL. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. J Polym Sci Part C Polym Symp. 1964;6:183–95.
Starink MJ. The determination of activation energy from linear heating rate experiments: a comparison of the accuracy of isoconversion methods. Thermochim Acta. 2003;404:163–76.
Dubaj T, Cibulková Z, Šimon P. An incremental isoconversional method for kinetic analysis based on the orthogonal distance regression. J Comput Chem. 2015;36:392–8.
Šimon P. Isoconversional methods. J Therm Anal Calorim. 2004;76:123–32.
Hou F, Martin JD, Dill ED, Folmer JCW, Josey AA. Transition zone theory of crystal growth and viscosity. Chem Mater. 2015;27:3526–32.
Dill ED, Josey AA, Folmer JCW, Hou F, Martin JD. Experimental determination of the crystallization phase-boundary velocity in the halozeotype CZX-1. Chem Mater. 2013;25:3932–40.
Dill ED, Folmer JCW, Martin JD. Crystal growth simulations to establish physically relevant kinetic parameters from the empirical Kolmogorov–Johnson–Mehl–Avrami model. Chem Mater. 2013;25:3941–51.
Hillis B, Losey B, Weng J, Ghaleb N, Hou F, Martin J. From rate measurements to mechanistic data for condensed matter reactions: a case study using the crystallization of [Zn(OH2)6][ZnCl4]. Crystals. 2016;7:11.
Kowalczyk M, Piorkowska E, Kulpinski P, Pracella M. Mechanical and thermal properties of PLA composites with cellulose nanofibers and standard size fibers. Compos Part Appl Sci Manuf. 2011;42:1509–14.
Supaphol P, Spruiell JE. Isothermal melt- and cold-crystallization kinetics and subsequent melting behavior in syndiotactic polypropylene: a differential scanning calorimetry study. Polymer. 2001;42:699–712.
Refaa Z, Boutaous M, Xin S, Siginer DA. Thermophysical analysis and modeling of the crystallization and melting behavior of PLA with talc: kinetics and crystalline structures. J Therm Anal Calorim. 2017;128:687–98.
Kalish JP, Aou K, Yang X, Hsu SL. Spectroscopic and thermal analyses of α′ and α crystalline forms of poly(l-lactic acid). Polymer. 2011;52:814–21.
Nelder JA, Mead R. A simplex method for function minimization. Comput J. 1965;7:308–13.
Wojdyr M. Fityk: a general-purpose peak fitting program. J Appl Crystallogr. 2010;43:1126–8.
Liao Y, Liu C, Coppola B, Barra G, Di Maio L, Incarnato L, et al. Effect of porosity and crystallinity on 3D printed PLA properties. Polymers. 2019;11:1487.
Müller AJ, Ávila M, Saenz G, Salazar J. CHAPTER 3. Crystallization of PLA-based materials. In: Jiménez A, Peltzer M, Ruseckaite R, editors. Polymer science, series. Cambridge: Royal Society of Chemistry; 2014, p. 66–98. http://ebook.rsc.org/?DOI=10.1039/9781782624806-00066.
Supaphol P, Spruiell JE, Lin J-S. Isothermal melt crystallization and melting behaviour of syndiotactic polypropylene. Polym Int. 2000;49:1473–82.
Acknowledgements
This research has been supported by the Spanish Ministry of Science and Innovation, MINECO Grant MTM2017–82724-R.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.

Rights and permissions
About this article

Cite this article
Díaz-Díaz, A.M., López-Beceiro, J., Li, Y. et al. Crystallization kinetics of a commercial poly(lactic acid) based on characteristic crystallization time and optimal crystallization temperature. J Therm Anal Calorim 145, 3125–3132 (2021). https://doi.org/10.1007/s10973-020-10081-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-10081-7