
Abstract
Nano-sized M0.05Eu0.05Ca0.9MoO4 (M=Li, Na, K, Rb, Cs) ceramics have been successfully synthesized by an aqueous sol–gel synthesis method using a tartaric acid as a ligand. In order to reveal the influence of the peculiarities of the nature of dopants effect into the crystallization of CaMoO4 double oxide, the thermal analysis of the as-prepared gels was performed. In addition, infrared spectroscopy was used in order to identify the functional groups from the characteristic stretching vibrations in the M–Eu–Ca–Mo–O tartrate gel precursors. Besides, to confirm the dynamics of growing crystallites in the final ceramics and to reveal the morphological changes on the surface, the x-ray diffraction and scanning electron microscopy were applied. Finally, photoluminescence measurements were used to estimate the optical properties of europium oxide as a dopant in the samples according to the nature of alkali metal. Therefore, according to the obtained results, it was estimated that luminescence intensity of Eu3+ ions is mainly affected by the chemical reaction, which takes place at about 973 K of temperature. This effect was partly confirmed from the results of the thermal decomposition of M–Eu–Ca–Mo–O tartrate gel precursors with an endothermic behaviour in the DSC curve, which indicates the crystallization mechanism of the CaMoO4 double oxide.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
CaMoO4 is a well-known and important material. For its unique chemical and optoelectrical properties, it has been used in various fields as a luminescent [1], optical fibre [2], magnetic [3] and solid-state laser [4] material. Additionally, it can be used in humidity sensors [5], photocatalysts [6], scintillators [7] and cryogenic detectors [8]. Predominantly, CaMoO4 has scheelite crystal structure [9] responsible for luminescence in blue and green parts of the spectrum [10], when excited with UV rays in 250–310 nm wavelength interval [11]. Calcium molybdate is also characterized by its strong physical and chemical resistances, which make it the preferred over its predecessors like Y2O2S:Eu3+, YVO4:Eu3+, Y2SiO5:Tb3+ or Gd2O2S:Tb3+ [12]. In this structure, the Mo atoms are bonded to four oxygen, forming the [13] clusters [14], which can absorb ultraviolet region with high absorption cross section [15]. Charge transfer between can be observed between [13] cluster and dopant ion [16], thus making it possible to achieve luminescence in different parts of the spectrum by doping CaMoO4 with rare-earth elements (RE). This peculiarity allows this material to be used as a matrix incorporated or saturated with dopant elements. By doping calcium molybdate with europium (III) ions (Eu3+), strong red luminescence is achieved [17], while doping with terbium (III) ions (Tb3+)—strong green luminescence [18, 19]. It was observed that doping CaMoO4 with two or more ions: rare-earth element (RE3+) and charge-compensating element, for example Bi3+ [20] or alkali metal (Li+, Na+, K+), luminescence intensity, is significantly improved without increasing RE3+ ion amount [21]. However, previously reported studies utilize inefficient or complex methods of synthesis, such as solid state or hydrothermal. Additionally, those methods require high annealing temperatures above or at 1173 K.
In recent years, microstructure and surface morphology have been proven to be essential part for achieving characterizing desired material properties [22]. Therefore, it is crucial to adequately choose a method and conditions for the synthesis of the desired material. There are several published methods, among which are: pulse laser ablation (PLA) [23], mixed oxide [24], molten salt [25], solid state [26], hydrothermal and its variations [13, 27, 28], Czochralski [29] and precipitation [30], but generally, scheelite-type phosphors are synthesized by conventional solid-state reaction method [31]. This approach usually requires annealing at high temperatures for several hours and subsequent grinding. This crushing process changes the phosphor surfaces, resulting in the loss of emission intensity. Therefore, demand for alternative synthesis methods such as hydrothermal method or sol–gel process arise, as it was also shown that phosphors prepared via an wet chemical routes have higher uniformity in particle-size distribution with high crystallinity and exhibit higher photoluminescence intensity than those prepared by the solid-state reaction [32]. Therefore, sol–gel is an important method for its simplicity, repeatability, mild synthesis conditions, potential industrial application and desirable achieved product morphology.
Moreover, we provide a simple aqueous route for synthesizing high-purity scheelite CaMoO4 phase substituted with RE element and a charge-compensating alkali ion. To our knowledge, such scale research was not published yet, as most of the published material presents solid-state or hydrothermal synthesis, and none of it compares the effect of all the substituted alkali ions (Li, Na, K, Rb, Cs). Additionally, thermal characterization of these materials is very scarce. While previous materials used Eu(III) ion hosts have been reported to have poor thermal stability [33], similar structures, such as Ho3+/Tm3+/Yb3+-codoped CaWO4, have been reported to have very high thermal stability, with only phase transition/organic impurity decomposition below 773 K after which the mass remained constant [34]. On the other hand, it gives this work an extra novelty and relevance, because of this synthesis technique that is based on the use of tartaric acid as a ligand during the sol–gel processing, which is powerful enough to prepare pure calcium molybdate with stabile crystal structure, uniform surface morphology and identical properties [35, 36]. Besides, our proposed synthesis procedure does not have any side effects to the final ceramic product and there are no typical undesirable consequences for the environment because of uses of tartaric acid that is found in food industry. According to that, it is obvious that this research enables to estimate the influence of the nature of alkali metal to the luminescent properties of Eu3+ ion in the host of CaMoO4.
Materials and methods
The samples of M0.05Eu0.05 Ca0.9MoO4 (M=Li, Na, K, Rb, Cs) were prepared by the tartaric acid-assisted synthesis (TAS) approach during the mixing stage of the initial salts employing an tartaric acid as a chelating agent that interacts as a ligand at the molecular level with the reaction mixture during the both dissolution in water and sol–gel formation.
Lithium (I) nitrate (LiNO3, 99% Alfa Aesar), sodium (I) nitrate (NaNO3 99,5% Alfa Aesar), potassium (I) nitrate (KNO3, 99,5% Alfa Aesar), rubidium (I) nitrate (RbNO3 99.9% Alfa Aesar), caesium (I) nitrate (CsNO3, 99,8% Alfa Aesar), europium (III) oxide (Eu2O3, 99.99% Alfa Aesar), calcium (II) nitrate tetrahydrate (Ca(NO3)2·4H2O 99% Alfa Aesar) and molybdenum (VI) oxide (MoO3, 99.95% Alfa Aesar) were used as starting materials and weighed according to the desired stoichiometric ratio (Table 1).
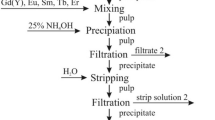
Nitric acid (HNO3 66% Reachem), distilled water and concentrated ammonia solution (NH3·H2O 25% Penta) were used as solvents and reagents to regulate the pH of the solutions. Tartaric acid (L–(+)–Tartaric acid (C4H6O6) (TA) ≥ 99.5% Sigma-Aldrich) was applied for escalation of solubility via coordination of starting compounds in the reaction mixture, especially, during the pH changes and evaporation before sol–gel formation. The synthesis scheme for the preparation of M–Eu–Ca–Mo–O tartrate gel precursors is shown in Fig. 1.

Synthesis scheme of the sol–gel preparation for the M–Eu–Ca–Mo–O tartrate gel precursors and M0.05Eu0.05Ca0.9MoO4 (M=Li, Na, K, Rb, Cs) ceramics
At first, MoO3 was dissolved in a solution that consisted of 25 mL of distilled water and 1 mL of concentrated ammonia by stirring at 355–360 K of temperature. Then, after continuous stirring at approximately 355–360 K as long as the excess of ammonia had been evaporated, the tartaric acid was added to the reaction mixture. The amount of tartaric acid is directly related to the concentration of molybdenum oxide according to their molar ratio of Mo/TA = 0.25. After the dissolution of tartaric acid in the reaction mixture, the required amount of alkali metal nitrate was added. Then, under the continuous stirring and heating, the europium (III) oxide and concentrated nitric acid were poured to the beaker. A clear yellowish solution was obtained and subsequently concentrated by slowly vaporizing the reaction mixture at 360 K.
In the following stage, a yellow transparent sol was formed after nearly 95% of the water had been evaporated. After drying the sol precursor in an oven at 393 K, the fine-grained light brown gel powders were obtained. The well-dried gel powders were used for the thermal analysis in order to estimate their decomposition mechanism. Meanwhile, the residue of gel precursors was additionally heat-treated at the temperatures of 623 K, 673 K, 773 K, 873 K, 973 K, 1073 K, 1223 K and 1273 K for 5 h in the air.
Thermal measurements were taken with TG‒DSC, STA 6000 PerkinElmer instrument using a sample weight of about 10 mg and a heating rate of 20° min−1 under the continuous air flow (20 cm3 min−1) at ambient pressure from room temperature to 1223 K. The Fourier transform infrared (FT-IR) spectra for the M–Eu–Ca–Mo–O tartrate gel precursors were recorded with a PerkinElmer Frontier FT-IR spectrometer. The corresponding gel precursors heat-treated at different temperatures, and the residual powders that were left after the thermal treatment were additionally investigated by x-ray diffraction (XRD) analysis. The XRD patterns were recorded in air at room temperature with a powder x-ray diffract meter Rigaku MiniFlex II using CuKα1 radiation. The patterns were recorded at the standard rate of 1.5 2θ/min−1. In addition, the Rietveld refinement of the obtained XRD patterns was performed using X’Pert HighScore Plus software. The scanning electron microscope (SEM) Hitachi SU-70 was used to study the surface morphology and microstructure of the obtained ceramic samples. Finally, the measurements of photoluminescence (PL) and photoluminescence excitation (PLE) spectra were taken by using a PerkinElmer LS-55 spectrometer.
Results and discussion
Thermal analysis
The TG/DTG/DSC measurements of the M–Eu–Ca‒Mo–O tartrate (M=Li, Na, K, Rb, and Cs) gel precursors, which were taken up to 1223 K at a heating rate of 20° min−1 in flowing air (20 cm3 min−1) at ambient pressure with a sample mass of about 10 mg are shown in Fig. 2, respectively. Tables 2–6 show data, which were deducted from Fig. 2 and supplements quantitatively the general view of thermal decomposition processes in M‒Eu–Ca‒Mo–O tartrate gel precursors.

Combined TG‒DTG‒DSC curves of the Li–Eu–Ca‒Mo–O tartrate gel precursor for Li0.05Eu0.05Ca0.9MoO4 (a), Na0.05Eu0.05Ca0.9MoO4 (b), K0.05Eu0.05Ca0.9MoO4 (c), Rb0.05Eu0.05Ca0.9MoO4 (d) and Cs0.05Eu0.05Ca0.9MoO4 (e) ceramics
In addition, it is also important to note that in the frame of this study a new investigation approach to the analysis of the thermal processes was applied during which it is possible to reveal and to calculate the overlapping chemical and physical changes. In this case, the nature of alkali metal plays a very important role, taking into account that all synthesis procedures were made in similar manner, and the way of the thermal decomposition of initial gels was not identical. These discrepancies allow us to estimate the influence of both the initial composition to the crystallization for the final ceramic materials and thermal decomposition of intermediate organic parts from the gel precursors. Especially, an important role goes to the exothermic effect in the DSC curve between 400 and 450 K of temperature, which thermal behaviour tends to increase by changing the nature of alkali metal in the sample. This exothermic effect was identified from the slope of DTG curves, which gradually converts into the peak by changing the nature of alkali metal in the initial composition. In our previous report, [37] we suggested a stage between the temperature of 421 K and 493 K assign to the thermal decomposition of tartaric-acids dimer (C8H8O10) into carbon monoxide (CO), carbon dioxide (CO2) and two acetic acid (CH3COOH) molecules. Meanwhile, by investigating this case, it is clear that the decomposition effect of tartaric-acids dimer is not very well pronounced and this process is closely related to the evaporation and removal of volatile components from the M–Eu–Ca‒Mo–O tartrate gel. Besides, an enhanced ionic character of the alkali metal reduces the coordination of unreacted tartaric acid molecules, for that reason their decomposition into the carbon oxides occurs at much lower temperature than was expected. Nevertheless, the decomposition of unreacted tartaric acid occurs up to 568 K in all cases with the mass loss of about 36–43%. The mass of the gel precursors during this decomposition stage, in the range of temperature from 300 K to 568 K, decreases by 36–37%, and this is typical for Li0.05Eu0.05 Ca0.9MoO4 and Na0.05Eu0.05 Ca0.9MoO4 initial composition compounds. Meanwhile, the mass change for K‒Eu–Ca‒Mo–O and Rb‒Eu–Ca‒Mo–O tartrate gel precursors increases to 40.261% and 41.176%, respectively, indicating enhanced ionic character of corresponding alkali metals. Moreover, the thermal behaviour of DSC curve clearly shows several heat changes, which are less defined in the range from about 450 K to 550 K of temperature. Finally, the last, fifth case is characterized by the biggest mass change, which is equal to 43.323%; however, the DSC curve has similar behaviour to those cases, which are shown in Fig. 2a, b.
As shown in Tables 2–6, stage II corresponds to the decomposition of metal tartrates. The mass of the gel precursors during this decomposition stage, in the range of temperature from 563 K to 803 K, decreases by 18.8–22.7%, except Li0.05Eu0.05Ca0.9MoO4 case, when the mass loss of about 20.2% ends at 837.5 K. The heat-treatment temperature of about 573 K is important because of the beginning of crystallization for the final CaMoO4 double oxide. Furthermore, this endothermic process is competing with gradual decomposition of metal tartrates, which takes place until the partial stabilization of TG curve at about 873 K of temperature. By further increasing the heating temperature from 823 to 973 K, the mass change of the samples varies from 11.1 to 12.9% and is directly related to the burning of carbon-based residue, which is formed from the tartaric acid and metal tartrates at lower temperatures. Finally, the last mass change of the M‒Eu–Ca‒Mo–O tartrate gel precursors in the range of temperature from 1047 to 1223 K does not exceed 0.1%, and, except endothermic behaviour because of further growth of crystalline phases, no significant changes in the DSC curve were identified. Besides, it is important to note that in all cases, except for Li0.05Eu0.05Ca0.9MoO4 ceramic, the tendency of slight mass changes above 973 K was observed. The main reason of these changes is directly related to the partial releasing and evaporation of molybdenum oxide from the multicomponent oxide.
In order to show both the decomposition process of tartaric acid in the gel precursors and the overall mass change peculiarities of volatile components in the gels according to the gas atmosphere, the thermal analysis of as-prepared samples was also performed under inert flow. In this case, the TG/DTG/DSC measurements of the M–Eu–Ca‒Mo–O tartrate (M=Li, Na, K, Rb, and Cs) gel precursors, which were carried out up to 1200 K at a heating rate of 20° min−1 in flowing nitrogen (20 cm3 min−1) at ambient pressure with a sample mass of about 5 mg, are shown in Fig. 3, respectively.

Combined TG‒DTG‒DSC curves of the Li–Eu–Ca‒Mo–O tartrate gel precursor for Li0.05Eu0.05Ca0.9MoO4 (a), Na0.05Eu0.05Ca0.9MoO4 (b), K0.05Eu0.05Ca0.9MoO4 (c), Rb0.05Eu0.05Ca0.9MoO4 (d) and Cs0.05Eu0.05Ca0.9MoO4 (e) ceramics. The overall view of the curves is shown on the right, and the highlighted temperature range from 303 to 770 K is presented on the left
From these results, one is clear that the decomposition stages of tartaric acid only slightly depend according to the heating atmosphere. The heating range in case of inert gas flow from 303 to 770 K (Fig. 3 left side) is almost identical compared with the cases presented in Fig. 2. Moreover, the behaviour of TG curves remains the same up to 873 K of temperature, and this concludes about similar heat-treatment effects, which are confirmed by DSC curves. Finally, the last mass change of the M–Eu–Ca–Mo–O tartrate gel precursors in the range of temperature from 873 to 950 K is attributed to the decomposition of a mixture from carbon and oxygen atoms, which are composed from the gel residue. This effect is confirmed by the results obtained from the K–Eu–Ca–Mo–O sample, where the behaviour of TG and DSC curves reiterates only in a more stretched range of temperature compared with the measurement in air flow.
In conclusion, it is also important to note that the total mass loss of all gel precursors well matches with the results obtained in an air atmosphere, while a prior mass change for the K–Eu–Ca–Mo–O sample above 873 K of temperature has no sufficient influence on the results, which were obtained in other heating conditions. Therefore, such an action is important only in the inert atmosphere and it is determined by the stability of final ceramics in a reducing environment. In this case, the combustion process in the air at temperatures above 873 K eliminates any specific conditions created by elemental carbon present in the sample.
FT-IR analysis
In order to show the characteristic group frequencies for the gel precursors in the mid-infrared region, the infrared spectroscopy (FT-IR) was performed. Corresponding FT-IR spectra are shown in Fig. 4, respectively.

FT-IR transmittance spectra of the M–Eu–Ca–Mo–O tartrate gel precursors dried at 393 K of temperature
According to the initial composition of the obtained M–Eu–Ca–Mo–O tartrate gel precursors, it is obvious that the stretching bands, which belong to the functional group frequencies of tartaric acid, in the infrared spectra are expected. It is well known that carboxylic acids (RCOOH) exist as dimers due to strong intermolecular hydrogen bonding. These compounds show a strong broad O–H stretching band in the 3300–2500 cm−1 range. The C=O stretching band of the dimer is observed near 1700 cm−1, while the free acid band is observed at higher wavenumbers (1760 cm−1). In addition, carboxylic acids show characteristic C–O stretching and in-plane and out-of-plane O–H bending bands at 1240, 1430 and 930 cm−1, respectively [38].
The range from 1660 to 1620 cm−1 is characteristic to the nitrate NO2 asymmetric stretching. This band overlaps with the intensive stretching of the C=O group at 1700 cm−1 in the carboxylic acids. Moreover, the weak band of nitrate NO2 symmetric stretching in the range from 1300 to 1270 am−1 is also observed. In addition, barely visible characteristic peak of nitrate N–O stretching in the range from 870 to 840 cm−1 was detected. Meanwhile, the characteristic stretching for the nitrate NO2 bending (710–690 cm−1) was not found [38]. Such a result explains the emergence of a peak in the DSC curve at 413.37 K, which tends to increase from the Li–Eu–Ca–Mo–O to Cs–Eu–Ca–Mo–O sample. This exothermal effect is closely related to the decomposition processes of the nitrate residue that left in the gel precursors after the evaporation procedure. The amount of corresponding nitrates in the gel precursors increases negligible and slightly influences the decomposition of the tartaric-acids dimer in the range of temperature from 416.55 to 455.48 K.
The characteristic infrared bands of five-atom inorganic molecules, as well as for CrO2−4 group, commonly are at the 890 cm−1 and 378 cm−1 of wavenumber. In all cases, the band at about 890 cm−1 observed in the FT-IR spectra, which is attributed to the vibrations of MoO2−4 functional group [38]. Moreover, the vibrational frequencies of tetrahedral MoO2−4-type compound for their M–O vibrations are observed at 897 cm−1 (ν1) and 837 cm−1 (ν3), respectively [39]. Finally, the metal–oxygen characteristic vibrations similar to those observed in the crystalline Powellite structure of CaMoO4 are attributed to the range from 800 to 500 cm−1 [40].
X-ray diffraction
The powder x-ray diffraction (XRD) patterns of the Li–Eu–Ca‒Mo–O tartrate gel precursor heat-treated at different temperatures shown in the top nine panels of Fig. 5 are well matched with the standard ICSD card of CaMoO4 that is presented in the bottom panel.

Standard ICSD card of CaMoO4 and XRD patterns of the Li–Eu–Ca‒Mo–O tartrate gel precursor dried at 393 K for 24 h and heat-treated at temperatures of 623 K, 673 K, 773 K, 873 K, 973 K, 1073 K, 1173 K and 1273 K for 5 h in air
No characteristic peaks attributable to the impurities by increasing heat-treatment temperature from 623 to 1273 K were identified. In addition, it is important to mention that drying of Li–Eu–Ca‒Mo–O tartrate gel precursor for Li0.05Eu0.05Ca0.9MoO4 ceramic at 393 K showed fully amorphous character and no characteristic peaks to any crystalline compound from the XRD pattern were found. Besides, these results are in a good agreement with the TG‒DTG‒DSC data, when the crystallization of the final tetragonal phase below 623 K of temperature was estimated. Moreover, an important conclusion was made, by comparing these analysis techniques that the crystallization of the final ceramic starts from about 568 K immediately after the initial decomposition of unreacted tartaric acid molecules into the elemental carbon and carbon oxides. According to above subject, the experimental and analysis results of other samples showed almost identical peculiarities. The end of the initial decomposition of tartaric acid for Li0.05Eu0.05Ca0.9MoO4, Na0.05Eu0.05Ca0.9MoO4, K0.05Eu0.05Ca0.9MoO4, Rb0.05Eu0.05Ca0.9MoO4 ceramics was similar and equal to 566 K of temperature, respectively. Meanwhile, the start of crystallization for Cs0.05Eu0.05Ca0.9MoO4 compound began at slightly lower 561 K of temperature. This effect was assigned to the increased ionic character of caesium, which plays a much more important role during the sol–gel formation stage, comparing it with other cases.
In order to prove that the crystallization of the final ceramics plays crucial role not only for the thermal decomposition of metal tartrates, but also strongly affects the physical properties, the Rietveld refinement analysis of XRD patterns for the synthesized multicomponent oxides was applied. The crystallite sizes and lattice parameter for the Li0.05Eu0.05Ca0.9MoO4, Na0.05Eu0.05Ca0.9MoO4, K0.05Eu0.05Ca0.9MoO4, Rb0.05Eu0.05Ca0.9MoO4, and Cs0.05Eu0.05Ca0.9MoO4 compounds heat-treated at different temperatures are shown in Table 7.
The dependency of the crystallite size versus temperature, collected from the Table 7, is shown in Fig. 6, respectively.

Plot of crystallite size versus heat-treatment temperature for Li0.05Eu0.05Ca0.9MoO4 (a), Na0.05Eu0.05Ca0.9MoO4 (b), K0.05Eu0.05Ca0.9MoO4 (c), Rb0.05Eu0.05Ca0.9MoO4 (d) and Cs0.05Eu0.05Ca0.9MoO4 (e) ceramics
From these curve graphs, it is clear that by increasing the sintering for M0.05Eu0.05Ca0.9MoO4 ceramics from 623 to 973 K, the values of crystallite size tend to increase; however, at somewhat higher temperatures their decreasing trends were observed. It is important to take attention to the fact that for Li0.05Eu0.05Ca0.9MoO4, Na0.05Eu0.05Ca0.9MoO4 and Cs0.05Eu0.05Ca0.9MoO4 ceramics, the decrease in the size for crystallites starts only above 1073 K of temperature. Meanwhile, the crystallites for K0.05Eu0.05Ca0.9MoO4 and Rb0.05Eu0.05Ca0.9MoO4 ceramics start to decrease already above 973 K. This effect is directly related to the crystallization of europium oxide [41], besides, depending on the nature of alkali metal in the ceramic, the growth of crystallites takes place at a different temperature.
SEM micrographs
According to such quite unexpected results obtained from the Rietveld refinement of XRD patterns, the surface morphology of K0.05Eu0.05Ca0.9MoO4 and Rb0.05Eu0.05Ca0.9MoO4 samples sintered at 1073 K for 5 h in air was also investigated. Corresponding SEM micrographs of these ceramics are shown in Fig. 7.

SEM micrographs of M–Eu–Ca‒Mo–O tartrate gel precursors for K0.05Eu0.05Ca0.9MoO4 (a) and Rb0.05Eu0.05Ca0.9MoO4 (b) ceramics heat-treated at 1073 K temperatures
As shown in Fig. 7a, the surface of the K–Eu–Ca‒Mo–O tartrate gel precursor for K0.05Eu0.05Ca0.9MoO4 ceramic heat-treated at 1073 K consists of spherical-like aggregated particles, which size varies from 200 nm to 1.2 µm. Meanwhile, the surface morphology of the Rb–Eu–Ca‒Mo–O tartrate gel precursor for Rb0.05Eu0.05Ca0.9MoO4 ceramic (Fig. 7b) consists of similar particles to those observed from the case shown in Fig. 7a, which sizes varies from 200 nm to 1.8 µm, respectively. In conclusion, it is clear that surface of both samples is almost identical and the difference that comes into existence is only related to the degree of either agglomeration or aggregation of individual particles.
PL analysis
In order to show the relation between the growth mechanism of the crystallites and luminescence properties of the ceramics prepared at 773 K of temperature, the photoluminescence (PL) measurement were applied. Corresponding PL excitation spectra of the M0,05Eu0,05Ca0,9MoO4 (where M=Li, Na, K, Rb, Cs) ceramics are shown in Fig. 8. The characteristic bands were registered with a fixed excitation at 395 nm of wavelength with emerged transitions from 7F0 → 5L6 to 7F0 → 5D2. This result matches well with UVLED and blue LED, respectively [42].

PL (λex = 395 nm) spectra of M–Eu–Ca‒Mo–O tartrate (M=Li, Na, K, Rb and Cs) gel precursors for M0.05Eu0.05Ca0.9MoO4 ceramics heat-treated at 773 K
The PLE spectra (Fig. 9) show characteristic 5D0 → 7FJ (J = 0, 1, 2, 3, 4) Eu3+ emission lines, which are summed up in Table 8 [43].

PLE (λem = 616 nm) spectra of M–Eu–Ca‒Mo–O tartrate (M=Li, Na, K, Rb and Cs) gel precursors for M0.05Eu0.05Ca0.9MoO4 ceramics heat-treated at 773 K
From the data displayed in the Table 8, it is clear that 5D0 → 7F3 transition is forbidden, which is observed only in Na0,05Eu0,05Ca0,9MoO4, Cs0,05Eu0,05Ca0,9MoO4, Rb0,05Eu0,05Ca0,9MoO4 samples, and such phenomena are explained only as J—mixing [43]. The characteristic emissions of Eu3+ from 5D1 are less common case; however, the peaks of 5D1 → 7F2 and 5D1 → 7F1 transitions were observed, which intensities are comparable with 5D0 → 7F1 characteristic emission. Meanwhile, the 5D0 → 7F0 transition is strictly forbidden according to the standard Judd–Ofelt theory; however, as was mentioned before, it could be explained by assuming that this transition is due to J—mixing [43]. Moreover, this transition is also an indicator of Cnv, Cn or Cs symmetries, because other symmetries do not produce observable spectral line. According to the results obtained in this work, a comparative study of the substitution effect on the intensity of luminescence by comparing intensity of 5D0 → 7F2 transition was also performed. Despite the fact that this transition of 4f configuration is hypersensitive to the symmetry and crystal field of Eu3+ ion, the ratio R of intensities for 5D0 → 7F2 and 5D0 → 7F1 characteristic emissions was used instead and expressed as I(5D0 → 7F2)/I(5D4 → 7F1), respectively. Figure 10 shows the before-mentioned ratio R distribution and its dependency from the crystallite size for M0,05Eu0,05Ca0,9MoO4 ceramics.

Plot of R versus crystallite size for M0.05Eu0.05Ca0.9MoO4 ceramics
From Fig. 10, it is clear that either growing or decreasing correlation cannot be observed. However, these results showed an interesting trend, which is directly related to both the sizes of crystallite that grew up at 773 K and the formation mechanism of obtained crystals at higher heat-treating temperatures. It is well known that smaller crystallites produce higher luminescence intensity. This is due to the fact that smaller crystallites would have less surface imperfections quenching luminescence and less energy would be wasted in bulk material as heat dissipation. On the other hand, this study also proved that further growth of crystallites at much higher temperatures completely differs according to the nature of alkali metal. This effect mainly depends from the distribution of metal salts in the gel, which formation, during the sol–gel processing, was influenced by interaction of individual ion. By the way, such conclusion shows that the optical properties of prepared ceramics are strongly affected by several factors, and the correlation is not always expected. Moreover, the tendencies of differences of both the physical properties and structural features for each ceramic sample remain even by increasing the heat-treating temperature.
Conclusions
Summarizing the results obtained from the TG/DTG/DSC, XRD, SEM and PL measurements, it can be concluded that the tartaric acid-assisted synthesis of M–Eu–Ca‒Mo–O tartrate (M=Li, Na, K, Rb, and Cs) gel precursors for nano-sized M0.05Eu0.05Ca0.9MoO4 ceramics is suitable preparation technique, which enables the detail investigation of multicomponent metal oxides in the frame of their chemical and physical properties. According to the thermal analysis, the decomposition of metal–tartrate gel precursors occurs in similar manner. Meanwhile, the emerging differences that mainly focus on the thermal decomposition of unreacted tartaric acid molecules, which mechanism depends on the coordination ability of the corresponding alkali metal. Besides, in all cases, except for Li0.05Eu0.05Ca0.9MoO4 ceramic, the tendency of slight mass changes above 973 K was observed. The FT-IR spectra of the M–Eu–Ca–Mo–O tartrate gels revealed that a small number of nitrates were left in the precursors. The XRD patterns correlated with the results of thermal analysis perfectly do not indicate any characteristic peaks attributable to the impurities by increasing the heat-treatment temperature from 623 to 1273 K. The dependency of the crystallite size versus temperature clearly showed that by increasing the heat-treating temperature from 623 to 973 K for M0.05Eu0.05Ca0.9MoO4 ceramics, the values of crystallite size tend to increase; however, at higher temperatures the decreasing trends of crystallites were observed. The reason of this behaviour directly depends from the start of the crystallization of europium (III) oxide. The study of surface morphology indicated the almost identical trends of morphological properties for the analyzed samples with the spherical particles, which size varies from 200 nm to 1.5 µm. The results of the PL studies revealed that the crystallite size and the mechanism of their growing tendencies for the M0.05Eu0.05Ca0.9MoO4 compounds are closely related to the ratio R of intensities for 5D0 → 7F2 and 5D0 → 7F1 characteristic emissions. Finally, it was concluded that the nature of alkali metal strongly affects the distribution of Eu2O3 in the CaMoO4 host and the reasons of such behaviour are basically determined by the different chemical activity of alkali metals in the range of temperature from 623 to 1073 K.
References
Hou ZY, Chai RT, Zhang ML, Zhang CM, Chong P, Xu ZH, et al. Fabrication and luminescence properties of one-dimensional CaMoO4:Ln3+ (Ln=Eu, Tb, Dy) Nanofibers via Electrospinning Process. Langmuir. 2009;25(20):12340–8.
Gao DJ, Lai X, Cui CH, Cheng P, Bi J, Lin DM. Oxidant-assisted preparation of CaMoO4 thin film using an irreversible galvanic cell method. Thin Solid Films. 2010;518(12):3151–5.
Lei F, Yan B. Hydrothermal synthesis and luminescence of CaMO4:RE3+ (M=W, Mo; RE=Eu, Tb) submicro-phosphors. J Solid State Chem. 2008;181(4):855–62.
Thongtem T, Kungwankunakorn S, Kuntalue B, Phuruangrat A, Thongtem S. Luminescence and absorbance of highly crystalline CaMoO4, SrMoO4, CaWO4 and SrWO4 nanoparticles synthesized by co-precipitation method at room temperature. J Alloy Compd. 2010;506(1):475–81.
Hosseinpour-Mashkani SS, Hosseinpour-Mashkani SS, Sobhani-Nasab A. Synthesis and characterization of rod-like CaMoO4 nanostructure via free surfactant sonochemical route and its photocatalytic application. J Mater Sci Mater Electron. 2016;27(5):4351–5.
Silva MMS, Sena MS, Lopes-Moriyama AL, Souza CP, Santos AG. Experimental planning of the synthesis of strontium molybdate by EDTA-citrate and its structural influence, morphology and optical bandgap. Ceram Int. 2018;44(14):16606–14.
Sun Y, Ma JF, Jiang XH, Fang JR, Song ZW, Gao C, et al. Ethylene glycol-assisted electrochemical synthesis of CaMoO4 crystallites with different morphology and their luminescent properties. Solid State Sci. 2010;12(7):1283–6.
Parchur AK, Ningthoujam RS, Rai SB, Okram GS, Singh RA, Tyagi M, et al. Luminescence properties of Eu3+ doped CaMoO4 nanoparticles. Dalton Trans. 2011;40(29):7595–601.
Wang YG, Ma JF, Tao JT, Zhu XY, Zhou J, Zhao ZQ, et al. Low temperature synthesis of CaMoO4 nanoparticles. Ceram Int. 2007;33(4):693–5.
Raju GSR, Pavitra E, Ko YH, Yu JS. A facile and efficient strategy for the preparation of stable CaMoO4 spherulites using ammonium molybdate as a molybdenum source and their excitation induced tunable luminescent properties for optical applications. J Mater Chem. 2012;22(31):15562–9.
Li X, Yang ZP, Guan L, Guo JX, Wang Y, Guo QL. Synthesis and luminescent properties of CaMoO4:Tb3+, R + (Li + , Na + , K +). J Alloy Compd. 2009;478(1–2):684–6.
Zhang ZJ, Chen HH, Yang XX, Zhao JT. Preparation and luminescent properties of Eu3+ and Tb3+ ions in the host of CaMoO4. Mater Sci Eng B Solid State Mater Adv Technol. 2007;145(1–3):34–40.
Luo YS, Dai XJ, Zhang WD, Yang Y, Sun CQ, Fu SY. Controllable synthesis and luminescent properties of novel erythrocyte-like CaMoO4 hierarchical nanostructures via a simple surfactant-free hydrothermal route. Dalton Trans. 2010;39(9):2226–31.
Marques VS, Cavalcante LS, Sczancoski JC, Alcantara AFP, Orlandi MO, Moraes E, et al. Effect of different solvent ratios (water/ethylene glycol) on the growth process of CaMoO4 crystals and their optical properties. Cryst Growth Des. 2010;10(11):4752–68.
Parchur AK, Ningthoujam RS. Preparation and structure refinement of Eu3+ doped CaMoO4 nanoparticles. Dalton Trans. 2011;40(29):7590–4.
Dutta S, Som S, Sharma SK. Luminescence and photometric characterization of K+ compensated CaMoO4:Dy3+ nanophosphors. Dalton Trans. 2013;42(26):9654–61.
Haque MM, Kim DK. Luminescent properties of Eu3+ activated MLa2(MoO4)(4) based (M=Ba, Sr and Ca) novel red-emitting phosphors. Mater Lett. 2009;63(9–10):793–6.
Cavalli E, Boutinaud P, Mahiou R, Bettinelli M, Dorenbos P. Luminescence dynamics in Tb3+-doped CaWO4 and CaMoO4 crystals. Inorg Chem. 2010;49(11):4916–21.
Parchur AK, Prasad AI, Ansari AA, Rai SB, Ningthoujam RS. Luminescence properties of Tb3+-doped CaMoO4 nanoparticles: annealing effect, polar medium dispersible, polymer film and core-shell formation. Dalton Trans. 2012;41(36):11032–45.
Zhang JH, Wang L, Jin Y, Zhang X, Hao ZD, Wang XJ. Energy transfer in Y3Al5O12:Ce3+, Pr3+ and CaMoO4:Sm3+, Eu3+ phosphors. J Lumin. 2011;131(3):429–32.
Liu XG, Li L, Noh HM, Jeong JH, Jang K, Shin DS. Controllable synthesis of uniform CaMoO4:Eu3+, M+(M=Li, Na, K) microspheres and optimum luminescence properties. RSC Adv. 2015;5(13):9441–54.
Laguna M, Nunez NO, Becerro AI, Ocana M. Morphology control of uniform CaMoO4 microarchitectures and development of white light emitting phosphors by Ln doping (Ln=Dy3+, Eu3+). CrystEngComm. 2017;19(12):1590–600.
Cho K, Choi J, Kim KM, Kim TW, Lee JI, Ryu JH. Pulsed laser synthesis of Er3+/Yb3+Co-doped CaMoO4 colloidal nanocrystal and its upconversion luminescence. J Nanosci Nanotechnol. 2016;16(6):6344–9.
Choi GK, Kim JR, Yoon SH, Hong KS. Microwave dielectric properties of scheelite (A=Ca, Sr, Ba) and wolframite (A=Mg, Zn, Mn) AMoO(4) compounds. J Eur Ceram Soc. 2007;27(8–9):3063–7.
Yan SX, Zhang JH, Zhang X, Lu SZ, Ren XG, Nie ZG, et al. Enhanced red emission in CaMoO4:Bi3+, Eu3+. J Phys Chem C. 2007;111(35):13256–60.
Parhi P, Singh SS, Ray AR, Ramanan A. Mechanochemically assisted room temperature solid state metathesis reaction for the synthesis of MMoO4 (M=Ca, Sr and Ba). Bull Mat Sci. 2006;29(2):115–8.
Yu S, Lin ZB, Zhang LZ, Wang GF. Preparation of monodispersed Eu3+:CaMoO4 nanocrystals with single quasihexagon. Cryst Growth Des. 2007;7(12):2397–9.
Yoon JW, Ryu JH, Shim KB. Photoluminescence in nanocrystalline MMoO4 (M=Ca, Ba) synthesized by a polymerized complex method. Mater Sci Eng B Solid State Mater Adv Technol. 2006;127(2–3):154–8.
Wangkhem R, Yaba T, Singh NS, Ningthoujam RS. Red emission enhancement from CaMoO4:Eu3+ by co-doping of Bi3+ for near UV/blue LED pumped white pcLEDs: energy transfer studies near. J Appl Phys. 2018;123(12):10.
Yin YK, Gao Y, Sun YZ, Zhou BB, Ma L, Wu X, et al. Synthesis and photoluminescent properties of CaMoO4 nanostructures at room temperature. Mater Lett. 2010;64(5):602–4.
Shi S, Gao J, Zhou J. Effects of charge compensation on the luminescence behavior of Eu3+ activated CaWO4 phosphor. Opt Mater. 2008;30(10):1616–20.
Kim KM, Ryu JH, Mhin SW, Park GS, Shim KB. Luminescence of nanocrystalline Tb(3)Al(5)O(12): Ce(3+) phosphors synthesized by nitrate-citrate gel combustion method. J Electrochem Soc. 2008;155(10):J293–6.
Lian JB, Qin H, Liang P, Liu F. Co-precipitation synthesis of Y2O2SO4:Eu3+ nanophosphor and comparison of photoluminescence properties with Y2O2:Eu3+ and Y2O2S:Eu3+ nanophosphors. Solid State Sci. 2015;48:147–54.
Cho H, Hwang SM, Bin Lee J, Ka DH, Kim TW, Lee BS, et al. White luminescence of Ho3+/Tm3+/Yb3+-codoped CaWO4 synthesized via citrate complex route assisted by microwave irradiation. Trans Nonferrous Met Soc China. 2014;24:S134–40.
Braziulis G, Janulevicius G, Stankeviciute R, Zalga A. Aqueous sol-gel synthesis and thermoanalytical study of the alkaline earth molybdate precursors. J Therm Anal Calorim. 2014;118(2):613–21.
Braziulis G, Stankeviciute R, Zalga A. Sol–gel derived europium doped CaMoO4:Eu3+ with complex microstructural and optical properties. Mater Sci Medzg. 2014;20(1):90–6.
Zalga A, Gaidamaviciene GE, Gricius Z, Uzpurvyte E, Gadeikis J, Diktanaite A, et al. Aqueous sol-gel synthesis, thermoanalytical study and electrical properties of La2Mo2O9. J Therm Anal Calorim. 2018;132(3):1499–511.
Stuart B. Infrared spectroscopy: fundamentals and applications. West Sussex: Wiley; 2004.
Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. Handbook of Vibrational Spectroscopy. 2006.
Braziulis G, Stankeviciute R, Zalga A. Sol-gel derived europium doped CaMoO4:Eu3+ with complex microstructural and optical properties. Mater Sci. 2014;20(1):90–6.
Curtis C, Tharp A. Ceramic properties of europium oxide. J Am Ceram Soc. 1959;42(3):151–6.
Liu J, Lian HZ, Shi CS. Improved optical photoluminescence by charge compensation in the phosphor system CaMoO4:Eu3+. Opt Mater. 2007;29(12):1591–4.
Binnemans K. Interpretation of europium(III) spectra. Coord Chem Rev. 2015;295:1–45.
Acknowledgements
This research was funded by the European Social Fund under the No 09.3.3-LMTK-712 “Development of Competences of Scientists, other Researchers and Students through Practical Research Activities” measure.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gaidamavičienė, G., Janulevičius, G., Venslauskaitė, E. et al. Aqueous sol–gel synthesis, thermoanalytical study and luminescent properties of M0.05Eu0.05Ca0.9MoO4 (M=Li, Na, K, Rb, Cs) nanocrystallites. J Therm Anal Calorim 140, 2185–2201 (2020). https://doi.org/10.1007/s10973-019-08962-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08962-7