
Abstract
Nano- and micro-sized La2Mo2O9 ceramics have been successfully synthesized by an aqueous sol–gel synthesis method using a tartaric acid as a ligand. Thermal analysis of both as-prepared La–Mo–O tartrate gel precursor and La2Mo2O9 double oxide heat-treated at different temperatures was performed. The high-temperature phase transition from α to β-phases was additionally investigated by broadband high-temperature impedance spectroscopy techniques. The α ↔ β phase transition was characterized by monitoring the conductivity and a transfer of heat with respect to temperature. The crystalline phase stability of monoclinic La2Mo2O9 compound was confirmed by x-ray diffraction measurements, while the dense nanoscale surface composed of the particles, ranging from 50 to 100 μm, was confirmed by scanning electron microscopy.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Multicomponent crystalline oxide materials that exhibit a high electrical conductivity associated with the phase transitions and an anomalous mobility of oxide anions attract both a fundamental and practical interest, in particular, owing to the prospects of their use in many solid-state ionic devices, i.e., as a solid-state electrolyte in solid oxide fuel cells, oxygen pumps and membranes for oxygen separation, oxygen sensors, etc. [1, 2]. From this point of view, yttria-stabilized zirconia (YSZ) is an excellent solid electrolyte and commonly employed as an oxide ion conductor in technological applications [3]. Besides, several authors reported an ionic conductivity enhancement of the yttria-stabilized zirconia films and bulk materials when decreasing crystallite size to the nano- or sub-micrometer range [4,5,6,7,8,9]. Nevertheless, the contemporary enhancements to improve the ionic conductivity are not sufficient; therefore, there is a need for new oxide ion conductors that have higher ionic conductivity compared with YSZ at intermediate temperature. In recent years, the La2Mo2O9- type compounds, known as the LAMOX family, which was originally reported by Lacorre et al. [10], have attracted great interest due to their high oxygen-ion conductivity at medium temperatures [11]. La2Mo2O9 undergoes a reversible transition from monoclinic α to cubic β structure at ~ 853 K, leading to an increase in the ionic conductivity up to two orders of magnitude and reaching values higher than those corresponding to YSZ [12]. Depending on the rate of cooling of the sample after its preparation, La2Mo2O9 can exist at room temperature as a stable monoclinic α phase, metastable cubic βms phase, or a mixture of these phases [1]. Besides, the reasons how the temperature affects the transition from cubic β to monoclinic α phases are not fully clear. It is obvious that this phase transition of non-substituted La2Mo2O9 into the pseudo-monoclinic α-phase is detrimental for practical applications due to a drastic drop in the conductivity below 833 K and mechanical failure caused by the high thermal expansion between the high- and low-temperature polymorphs [13]. The possibilities of reducing the structure changes in La2Mo2O9 by the influence of temperature remain a great challenge in today’s science. In addition, the applicability of La2Mo2O9 is also limited due to its lower stability under reducing atmosphere, because of the presence of Mo6+, which is likely to join electrons by causing structure changes that strongly affects the conductivity as well as performance of used device [2]. Despite the above-mentioned drawbacks, La2Mo2O9 remains interesting because of its unique crystal structure and elemental composition, which allows the possibility of the substituting and doping that leads to the stabilization of high-temperature cubic phase and low decrease in conductivity at room temperature. For example, the substitution of molybdenum by tungsten significantly increases the resistance against the reduction environment impact, while replacing lanthanum side by Sr2+, Y3+, Zr4+ and Nb5+ ions stabilizes the β-phase and in some cases even increases the conductivity, which has an excellent characteristics only for high-temperature metastable cubic βms phase. Nevertheless, in order to explain in detail the possible influence of nonessential structural changes according to the substitution or addition, the further investigation and comparative study of non-substituted La2Mo2O9 ceramic by different techniques are required. It is also well known that the preparation technique has significant effect on the chemical and physical characteristics of final material; therefore, the relation of phase transition in the range of temperature between 783 and 853 K during the initial formation from starting compounds and reversible changes of annealed ceramic has additional motivation and is highly desirable.
Currently, there are various ways to synthesize LAMOX compounds. One of the most common is a solid-state route, where the initial ingredients are mixed and heated up to high temperatures [14]. However, this method does not allow to obtain directly dense ceramic samples, since a milling step is necessary before sintering. Indeed, even if this step leads to the decrease in the grain size and thus to a higher sinterability, it also leads to the apparition of impurities, especially when the milling balls/jar are made of zirconia. For instance, in 2011 a study was published claiming that La2Mo2O9 ceramic samples were obtained by solid-state synthesis in the temperature range of 1233–1373 K [15]. Other, more classical ways have been used, such as the citrate method [16], a Pechini or modified Pechini route [6, 17,18,19], the microwave-assisted method [20] and the EDTA complexation method [21]. Another popular way obtaining the sample of interest is a polyol process. The polyol process concerns the preparation of metallic powders by reduction of inorganic compounds in liquid polyols. A general reaction on model has been established with a reaction via the solution: dissolution of the solid precursor; reduction in solution; homogeneous nucleation and growth of the metallic phase from the solution [22].
From this point of view, in this work we have successfully synthesized La2Mo2O9 ceramic via rather rarely used aqueous tartaric acid-assisted sol–gel synthesis technique, which allowed to obtain monophasic crystalline compound at relatively low temperature of 823 K. In addition, it is important to note that the crystallization of high-temperature metastable cubic βms phase started from 783 K. Moreover, it was also showed that the increase in annealing temperature to 1473 K does not change the position of characteristic peaks for La2Mo2O9 ceramic as presented in the XRD patterns. These obtained results are very important for showing the highest capabilities of this synthesis technique, which was used in this work. The reason of selecting tartaric acid as a chelating agent lies within a couple of essential features that overcome some of the problems arising during the synthesis with citric acid. One of the improvements is the potential reduction of toxicity. This effect is justified regarding the study of thermal decomposition of a citric acid, which reveals that at 448 K, citric acid is converted into aconitic acid. Further heating results in formation of methyl maleic anhydride (III). Aconitic acid may also decompose to acetone [23], while the decomposition of tartaric acid mainly produces carbon dioxide and monoxide [24]. Also, citric acid has a higher molecular mass which causes a higher expansion of the gel precursor when it is being heat-treated to obtain a gel. During the sudden expansion, a part of the sample might be dropped out of the crucible. Finally, tartaric acid has been found to be a more effective chelating agent for LAMOX ceramics when the calcination is carried out at temperatures above 1323 K [25].
Experimental
The synthesis of La–Mo–O tartrate gel precursor for La2Mo2O9 ceramic was prepared by an aqueous sol–gel synthesis by using tartaric acid as a chelating agent that interacts as a ligand at the molecular level with the reaction mixture during the both dissolution in water and sol–gel formation. Lanthanum (III) oxide (La2O3, 99.99% Alfa Aesar) and molybdenum (VI) oxide (MoO3, 99.95% Alfa Aesar) were used as starting materials and weighed according to the desired stoichiometric ratio. Nitric acid (HNO3 66% Reachem), distilled water and concentrated ammonia solution (NH3 · H2O 25% Penta) were used as solvents and reagents to regulate the pH of the solutions. Tartaric acid (L–(+)–tartaric acid (C4H6O6) (TA) ≥ 99.5% Sigma-Aldrich) was applied for escalation of solubility via coordination of starting compounds in the reaction mixture, especially, during the pH changes and evaporation before sol–gel formation. At first, MoO3 was dissolved in a solution that consisted of 25 mL of distilled water and 0.5 mL of concentrated ammonia by stirring at 353–363 K temperature.
Then, after continuous stirring at approximately 353–363 K as long as the excess of ammonia had been evaporated, the tartaric acid with a molar ratio of Mo/TA = 0.25 was added during the continuous stirring to the reaction mixture at the same temperature.
Secondly, the lanthanum (III) oxide and concentrated nitric acid were added to the beaker. A clear solution was obtained and subsequently concentrated by slowly vaporizing the reaction mixture at 333 K.
In the following stage, a yellow transparent sol was formed after nearly 95% of the water had been evaporated under continuous stirring. After drying in an oven at 393 K, fine-grained dark blue gel powders were obtained.
Next to that, the La–Mo–O tartrate gel precursor for La2Mo2O9 ceramics were heat-treated for 5 h at 823, 873, 1073, and 1273 K. Finally, the powders were pelletized and ceramic sintered at 1473 K for 5 h in air. In addition, it is also important to note that all values related to the selection of heat treatment temperatures, duration time, and pressing conditions had been developed empirically by performing numerous tests to ensure the formation of a dense ceramics with no cracks or cavities.
The thermal analysis was performed with TG–DSC, STA 6000 Perkin-Elmer instrument using a sample mass of about 5 mg and a heating rate of either 20º min−1 for a gel precursor or 40º min−1 for sintered ceramics under airstream (20 cm3 min−1) at ambient pressure by raising the temperature up to 1223 K. The sample mass, heating rate, atmosphere and its flow rate were selected empirically during numerous tests to ensure the best signal peak efficiency and to minimize the noises and background signals, which occur because of the influence of corundum crucible and equipment limits. X-ray diffraction (XRD) patterns were recorded in air at room temperature by employing a powder x-ray diffractometer Rigaku MiniFlex II using Cu Kα1 radiation. The spectra were recorded at the standard rate of 1.5 2θ min−1. The sample was spread on the glass-holder in order to obtain maximum intensity of the characteristic peaks in the XRD pattern. The Rietveld refinement of the obtained XRD patterns was performed using X’Pert HighScore Plus software. The scanning electron microscopes (SEM) Hitachi TM3000 and Hitachi SU-70 were used to study the surface morphology and microstructure of the obtained ceramic samples. The measurements of electrical properties were performed using two different impedance spectroscopy techniques by newly developed impedance spectrometer [26, 27]. Sintered ceramics were processed, obtaining cylindrical samples of around 1.5 mm height and up to 3 mm diameter with Pt paste electrodes.
Results and discussion
Synthesis
The dissolution process of molybdenum (VI) oxide in water and ammonia solution is given in Eq. 1:
According to Ref. [28], the formation of [(MoO4)p(C4H4O6)qHr](2p+2q−r)− coordination compound is most likely, in this synthesis stage. Generally, the chemical reaction that occurred after this procedure can be written as shown in Eq. 2:
By slowly vaporizing the final reaction mixture, the chemical reactions that took place in this synthesis step are listed in Eqs. 3 and 4:
After drying in an oven, the dark blue gel powders were obtained, as illustrated in Eq. 5:
Thermal analysis
In this work, the thermal analysis as a powerful tool was properly used for both the detailed investigation of thermal decomposition of La–Mo–O tartrate gel precursor and the crystal phase transition from monoclinic α-phase to cubic β-phase for La2Mo2O9 ceramic. As can be seen in Fig. 1, the analysis of TG–DTG–DTA curves, which reveal the thermal decomposition of as-prepared La–Mo–O tartrate gel precursor, is rather complicated as expected, but one fact is clear, due to the relatively high amount of tartaric acid in the reaction mixture, the final mass loss of about 61% occurs up to 953 K. Further increase in heating temperature does not change the sample mass significantly, which gives the conclusion that the decomposition of all volatile organic parts in the gel precursor occurred below 953 K. Besides, it is also important to note that the sublimation of MoO3 does not start above 1073 K of temperature, which concludes about high stability of final crystalline compound. This result also confirms that above 1173 K the melting point of the final double oxide shifts to the higher temperatures, while in the case of La2Mo3O12 ceramic, as reported in our previous work, such behavior was not observed [29]. In addition, the mass of the sample powder tablet, sintered at 1473 K for 5 h in air atmosphere, has not changed and this fully satisfies the conclusion about the formation of really high stability of the final La2Mo2O9 ceramic. Table 1 shows the data, which were deducted from Fig. 1, and supplements quantitatively the general view of thermal decomposition processes in La–Mo–O tartrate gel precursor.

Combined TG–DTA curves of the La–Mo–O tartrate gel precursor for La2Mo2O9 ceramic
Therefore, the decomposition processes of the La–Mo–O tartrate system could be roughly divided into at least five stages that clearly indicate all mass losses and heat flow transformations occurring during the sample heating in the appropriate range of temperature. The first stage of the sample mass loss is closely related to the complete decomposition of residue tartaric acid, the excess of which has been added to reaction mixture in order to avoid precipitation during the gelation process. It is well known that the decomposition of tartaric acid usually occurs in three steps [24]. At first, tartaric acid loses one molecule of water and this dehydration of water molecules starts during the both final aqueous sol solution evaporating procedure on the hot plate and drying process in the oven at 393 K for 24 h in air atmosphere. It is also important to mention the fact that complete decomposition of nitric acid during the evaporating process allowed to improve the initial tartaric acid decomposition and also accelerated the dehydration of water molecules from the gel precursor. Besides, the temperature of hot plate was increased to 523 K, which enabled the tartaric acid removal process, before gel formation, in which the mass loss was of about 18.9%. This amount of tartaric acid loss was calculated taking in account the additives of starting compounds and residue of the sample mass from the reaction mixture before and after gelation process. According to the data presented in Table 1 and Eq. 6, the first mass loss of the as-prepared La–Mo–O gel precursor in stage I consists of 4.3% and this step may be formulated, as follows:
In the range of temperature from 303 to 420 K, a broad endothermic band with heat flow of 1078.1 mJ and ΔH = 106.7 J g−1 on the DSC curve corresponds to the first interval of the mass loss in the stage I. In addition, this combination of tartaric acids (TA) in the beginning of stage I, as presented in Eq. (6), is in a good agreement with the total amount (34.57%) of unreacted TA with molybdate ions (MoO42−), which mass loss contains of 34.5% (Table 1, Stage No. I, row 2).
By further increasing the temperature from 421 to 493 K, the decomposition of tartaric-acids dimer (C8H8O10) into carbon monoxide (CO), carbon dioxide (CO2) and two acetic acid (CH3COOH) molecules starts. The removal of these molecules from the gel is carried out simultaneously, and it is characterized by a broad endothermic band (110.6 mJ; ΔH = 11.0 J g−1), which attributed to the evaporation of the acetic acid from the sample. The exothermic behavior of the DSC curve at about 423 K of temperature is related to the removal of CO and CO2 molecules. Next to that, at somewhat higher temperatures, the dimer of tartaric acids already breaks down into two molecules of pyruvic acid (CH3COCOOH) and one molecule of carbon dioxide. Quantitatively, this decomposition process is characterized from 494 to 573 K of temperature and presented in Table 1 stage I row fourth. The last third endothermic band of stage I is attributed to the thermal evaporation of pyruvic acid; meanwhile, the exothermic behavior in the DSC curve is directly related to the releasing of carbon dioxide from the gel precursor. After the thermal decomposition of the free-tartaric acid in the La-Mo–O tartrate gel precursor with the mass change of 34.5%, the second stage of the partial breakdown of corresponding metal tartrates, as shown in Eq. 7, takes place:

As shown in Table 1 stage II row one, the initial decomposition of metal tartrates La10[(MoO4)10(C4H4O6)14]H18 begins with the removing of oxygen excess from the gel precursor. These oxygen ions are eliminated with the carbon and hydrogen atoms according to the amount ratios of carbon dioxide and water molecules, as seen from Eq. 7, respectively. The mass of the gel precursor during this decomposition stage, in the range of temperature from 574 to 793 K, decreases by 12%, and the transformation changes of this process could be divided into two different thermal effects. The first one, with endothermic character, starts at 574 K of temperature and is attributed to the evaporation of water molecules from the sample residue. Meanwhile, the other change of mass, from the 659 to 793 K of temperature, is related to well-expressed exothermic behavior in the DSC curve and is attributed to the releasing of carbon dioxide molecules from the residue of the gel precursor. In addition, it is also important to mention that after this range of temperature all hydrogen atoms were escaped in the form of water molecules. This is the main reason why after the second decomposition stage the changes of sample mass almost stopped and the crystallization of β-La2Mo2O9 cubic phase has started. This endothermic effect in the DSC curve is taking place from the 794 to 843 K, as shown in Fig. 1 and Table 1 stage III, respectively. This crystallization process is presented in Eq. 8:

It is also important to note that the heat consumption (569.5 mJ, ΔH = 5.2 J g−1) during the crystallization of metastable ceramic is similar to those values that were obtained and calculated according to the follow-up investigations of α ↔ β phase transition in the La2Mo2O9 powders.
Finally, the last mass change (13.3%) of the La–Mo–O tartrate gel precursor in the range of temperature from 844 to 953 K is attributed to burning of inorganic carbon, which is formed from the tartrates at lower temperatures. This combustion process is presented in Eq. 9 as follows:

This exothermic process is overlapped with both the growing of crystallites and the size of individual particles, which are characterized by the heat absorption. This is the reason why the symmetry of the exothermic peak in this range of temperature is not regular, and after abrupt increase is being observed the decrease with tendency into endothermic behavior. Before calculating the total release of the heat during the burning of organic residues in the sample, it is important to estimate the endothermic effects into the overall process. As seen in Table 1 stage IV row 1, the total heat is equal to the sum of exothermic and endothermic processes, which have occurred in the same time.
By further increasing the heating temperature from 954 to 1223 K, the mass change of the sample is extremely low, does not exceed 0.1%, and no significant changes in the DSC curve were identified. It means that after the sintering of the La–Mo–O tartrate gel precursor, the homogenous substance was formed with the thermal properties similar to those of La2Mo2O9 ceramic.
In order to confirm the phase transition at elevated temperatures in sintered La–Mo–O tartrate samples, their DSC measurements were performed. Table 2 shows the thermoanalytical data of the α → β phase transition for the La2Mo2O9 ceramics, which compare the influence of the sample mass and initial annealing temperature into the onset of this action.
The corresponding DSC curves of the La–Mo–O tartrate gel precursor, which was heat-treated at 873, 1273 and 1473 K temperatures, are shown in Fig. 2. From these results, it is clear that different surface morphology, which much depends on the heat treatment temperature, significantly affects both the onset temperature and the end temperature of the phase transition. Besides, it also influences the peak position, heat flow and enthalpy similarly as end temperature, which illustrate the state of monoclinic phase at room temperature and its relation to the surface properties.

DTA curves of the La2Mo2O9 ceramic heat-treated at 873, 1273, and 1473 K with different mass of the samples
Figure 3 displays the onset temperature of the phase transition of La2Mo2O9 ceramics as a function from sintering temperature. In this case, it is well seen that the initial mass, which was used for the measurement, slightly influenced the beginning of the phase transition from the monoclinic crystalline phase into metastable high-temperature cubic phase shown in the plot as the black curve and blue curve, respectively. The red dashed curve shows the linear dependency of the initial mass of LAMOX ceramic from the onset temperature, which is somewhere in the middle of analyzed samples. On the other hand, it is obvious that the onset temperature depends on the surface structure of the analyzed sample and this dependency is nonlinear.

The onset temperature of the phase transition of La2Mo2O9 ceramic as a function from sintering temperature
The dependency of the end temperature, peak position, heat flow and enthalpy from the sintering temperature of the LAMOX ceramic is shown in Figs. 4 and 5. From these curve graphs, it is clear that by increasing the sintering temperature of LAMOX ceramic from 873 to 1473 K the values of end temperature, peak position, heat flow and enthalpy tend to increase and at the higher temperatures start to decrease again. This phenomenon is explained by the two competing effects, like the surface area and degree of crystallinity. The LAMOX sample heat-treated at 873 K of temperature usually composed of particles with size from 100 to 200 nm, which is characterized by a large surface area. It means that in such conditions the area of grain boundary is either larger or similar to the surface, which is created by the particles itself. By increasing the thermal treatment, the surface area tends to decrease, but the degree of crystallinity increases. In other words, the surface area of grain boundary decreases, but the area of particles surface increases and this leads to the increase in energy of phase transition. From the temperature of 1273 K, the surface area decreases further, but the crystallinity increases much slower and this leads to the decreasing of energy of phase transition, as is shown in Figs. 4 and 5.

The end temperature and peak position of the phase transition of La2Mo2O9 ceramic as a function from sintering temperature

The heat flow and enthalpy of the phase transition of La2Mo2O9 ceramic as a function from sintering temperature
In order to show how this behavior of the onset temperature, end temperature, peak position, heat flow and enthalpy depends on the amount of monoclinic phase, which have formed after several heating and cooling procedures, the LAMOX ceramic was additionally investigated by DSC analysis, as shown in Figs. 6 and 7, respectively.

DTA curve of the La2Mo2O9 ceramic heat-treated at 873 K

DTA curve of the La2Mo2O9 ceramic heat-treated at 1473 K
From the first view, it is clear that the tendency of α → β phase transition for the La2Mo2O9 ceramic is similar to both samples heat-treated at 873 and 1273 K, respectively. The formation of cubic phase starts from about 843 K and ends at about 845 K. This stage was more or less similar in both cases during the additional thermal treatment, as seen from DSC results (Figs. 6 and 7). Meanwhile, the reversible phase transition from cubic to monoclinic phase started at lower temperature and its range was slightly different comparing the two cases mentioned above. In other words, the transformation of cubic phase starts from about 789 K and ends at 773 K for the case of LAMOX sample initially heat-treated at 873 K. Meanwhile, the initial formation of monoclinic phase in the range of temperature from 812 to 783 K was identified in the case of LAMOX sample initially heat-treated at 1473 K. This difference of reversible phase transition shows that the stability and surface structure of the ceramic heat-treated at 1473 K is better, because the heating range from 573 to 1073 K during the measurement slightly affects both the morphology and crystallinity of LAMOX sample that was heat-treated at 873 K.
Table 3 shows the thermoanalytical data of the α → β phase transition for the La2Mo2O9 ceramic under the influence of the sintering temperature and the amount of measurement. The graphical view of the data collected in Table 3 is presented in Figs. 8–12. From these results, one is clear that onset temperature of differently heat-treated samples after the first heating procedure tends to increase slightly from 826.84 to 830.32 K and during the second heating stage the opposite change direction of onset temperature from 829.39 to 828.22 K was observed. Such change in onset temperature is directly related to the transformations of surface morphology and the size of crystallites in the sample heat-treated at 873 K because of measurement that was performed in the range of temperature from 573 to 1073 K. This elevation of temperature made a lot of changes to the size of crystallites, which influenced the increase in Tonset from 826.84 to 829.39 K. In this case, almost identical effect was observed comparing the samples which were heat-treated at 873 and 1273 K (Fig. 3). Besides, after the second heating stage of the same sample the onset temperature only slightly decreased, this suggests only minimal changes in surface morphology and crystal structure, which were caused by phase transition during the cooling procedure.

The onset temperature change trend of the phase transition of La2Mo2O9 ceramic depending on the temperature of sintering
After the analysis of phase transition during the cooling the sample (Fig. 8), it is clear that the onset temperature tends to increase regardless of the amount of measurements. Nevertheless, the values of Tonset remain almost identical of both samples.
Similar behavior was observed analyzing the results from the values of end temperature and peak position during the cooling procedure, as shown in Figs. 9 and 10. It confirms that the phase transition from metastable cubic phase to monoclinic room temperature phase during the cooling of the sample is only little affected by the surface morphology. In this case, the values of onset temperature, end temperature and peak position increased only because of the change of crystallites, which tend to decrease during the cooling. On the other hand, though the values of end temperature and peak position during the heating procedure show both declining nature and increasing character, however, their numeric values are more or less similar comparing with the changes in onset temperature.

The end temperature change trend of the phase transition of La2Mo2O9 ceramic depending on the temperature of sintering

The peak position temperature change trend of the phase transition of La2Mo2O9 ceramic depending on the temperature of sintering
Figures 11 and 12 show the identical character of heat flow and enthalpy of both samples, which is expressed by Eq. 10.
where Q—heat flow [mJ], ΔH—enthalpy [J g−1]; and m—sample mass [mg].

The values of the heat flow change trend of the phase transition of La2Mo2O9 ceramic depending on the temperature of sintering

The values of the enthalpy change trend of the phase transition of La2Mo2O9 ceramic depending on the temperature of sintering
As shown in Fig. 3, the sample mass slightly influences the onset temperature and especially the heat that is released during the phase transition. The recalculation of heat into the enthalpy lets to avoid sample mass influence to the value of energy that is released or absorbed during the transformation of the explored crystal structure.
Figures 11 and 12 show almost identical dependency, because of really similar mass of the samples, which were used during the measurement. The heat and enthalpy values in the case of ceramic which was heat-treated at 873 K are similar independently from the amount of measurement. This result can be explained by the crystallization and structure rearrangement during the heating, because specific conditions were created when achieved relatively high 1073 K of temperature. In such case, the crystallization and morphological changes in the ceramic compensated each other and either heat or enthalpy remains similar. From this point of view, a completely different dependency was estimated in the sample that was heat-treated at 1473 K. Comparing with the ceramic that was prepared at 873 K, in this sample the heat as well as enthalpy tends to grow depending on the amount of measurements. This tendency is directly related only to the recrystallization processes during the phase transition in the final ceramic. The minimal surface changes, caused by its increased density, affect only really slightly on energy transformation in this case.
In conclusion, it is clear that the additional heating of obtained ceramic influences significantly the phase transition processes and could potentially affect the final physical properties expressed by ionic conductivity in this material. This is the main reason why further investigations of ionic conductivity of this system related to the crystallization and changes of surface morphology under elevated temperature must be explored.
X-ray diffraction
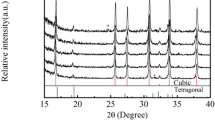
The powder x-ray diffraction (XRD) patterns of La–Mo–O tartrate gel precursor heat-treated at different temperatures shown in the top five panels of Fig. 13 are well matched with the standard ICSD card of La2Mo2O9 that is presented in the bottom panel. No characteristic peaks attributable to the impurities by increasing of sintering temperature from 823 to 1473 K were identified. These results led us to conclude about the high stability of obtained ceramic material, because no crystallization of the side crystal phases in whole sintering range was estimated. Besides, in contrast to our previous work with La2Mo3O12 [29], no melting of the LAMOX sample has not started above 1273 K. In addition, it is also interesting to mention that sintering of La–Mo–O tartrate gel precursor for La2Mo2O9 ceramic at 773 K showed fully amorphous character and no characteristic peaks of any crystalline compound from the XRD pattern were found. Besides, this result is in a good agreement with the TG–DSC data, when the crystallization of the final cubic phase from about 813 K of temperature was estimated.

Standard ICSD card of La2Mo2O9 and XRD patterns of the La–Mo–O tartrate gel precursor heat-treated at 823, 873, 1073, 1273, and 1473 K temperatures
In order to prove that the crystallization plays crucial role in phase transition for La2Mo2O9 ceramic, the Rietveld refinement analysis of all samples was also performed. The agreement indices and crystallite sizes for the La2Mo2O9 ceramic annealed at different temperatures are shown in Table 4.
From the data placed in Table 4, it is clear that by the increasing the sintering temperature the size of crystallites also increases. The crystallites size in the room temperature monoclinic phase is about 70 nm independently from the final sintering temperature above 1073 K of the final ceramic. It means that the investigations of phase transition of the samples heat-treated below 1073 K should be done with great care, because of growing of crystallites can dramatically affect the heat of phase transition. In addition, one may identify a formation of a pseudo-amorphic state in a sample, heat-treated at 1473 K. This kind of behavior could be clarified given the rather small surface area of the powder resulting from an insufficient amount of the sample being measured with x-ray diffractometer.
In conclusion, it is clear that the formation and crystallization of the final ceramics mainly depend on several factors like aqueous sol–gel processing conditions, sintering time and temperature.
SEM micrographs
In order to show both the crystal growth and densification of obtained ceramics by increasing sintering temperature, the surface morphology was investigated. Corresponding SEM micrographs of the La–Mo–O sample heat-treated at 823, 873, and 1473 K are shown in Fig. 14. From the x-ray diffraction studies, it is clear that the crystallization of the La2Mo2O9 ceramic starts at 823 K by the formation of well-agglomerated spherical particles in size of about 82–85 nm, as seen from both the above right micrograph (Fig. 14b) and the enlarge picture in Fig. 14a. In addition, there is well seen at least several layers of differently arranged spherical particles, which differs mostly by its density. In the less dense layer, the surface mainly composed of both well-agglomerated particles and the pores between them, diameter of which reaches more than 100 nm. Unlike porous layers, the surface of dense sheets mainly consists of well-aggregated spherical particles, size of which varies from 82 to 85.5 nm. As shown in the lower left part picture of Fig. 14c, by increasing the sintering temperature to 873 K the surface morphology of the analyzed sample has changed completely. In this case, due to thermal decomposition and melting of the aggregates and agglomerates the growing of both the particles and pores was observed. It is also important to note that the surface, in this case, is mainly composed of spherical and elliptical particles, size of which varies from 100 to 200 nm. Besides, the size of pores also increases and varies from 100 to 400 nm. The formation of porous surface morphology of the synthesized LAMOX sample is the critical stage due to the thermal treatment and can be a serious problem in order to obtain final dense-structured materials. This is the reason why additional measures like careful and meticulous powder grinding and pressing are needed. Therefore, the LAMOX powders heat-treated at the required temperature, well-grinded and properly pressed were additionally sintered at 1473 K. The results of obtained surface morphology and internal structure are shown in Fig. 14d, respectively. In this case, the formation of really dense structure with the circular shape crystals of size varying from 100 to 200 μm was achieved. Such surface morphology and single-crystal structure, confirmed by XRD, of the obtained LAMOX-type material is very important because of the interpretations of its electrical properties, which are done with great care. Moreover, the relatively large crystals with dense structure allow to eliminate all factors that usually significantly affect the physical properties of the sample.

SEM micrographs of La–Mo–O tartrate gel precursor for La2Mo2O9 ceramic heat-treated at a 823 K, b 823 K, c 873 K and d 1473 K temperatures
Impedance analysis
The typical Arrhenius plots of temperature dependencies of standard deviation for α and β crystal phase transition processes in La2Mo2O9 ceramic are shown in Fig. 15. According to these results, the ionic conductivity starts to increase by two orders from about 790 K and ends at about 831 K of temperature. These onset and end temperatures slightly differ compared to the values (Tonset = 829.27 K, Tend = 811.78 K), which were estimated using DSC analysis and are presented in Table 3. The resulting initial and final temperature differences can be explained by the different heating rate of the sample during measurement. In this case, the heating and cooling rates of the analyzed ceramic were chosen with great care and equal to 3 degree per minute. Despite this assumption, when the difference between onset temperatures reaches 40 degrees and the difference between end temperatures is equal to 20 degrees, only one conclusion is suggested that the formation of cubic beta-phase needs more energy and this process is less favourable than corresponding crystallization of monoclinic phase. Besides, the slightly higher activation energy of 1.09 eV of beta-La2Mo2O9 phase formation also confirms that cubic crystal structure is less stable comparing with monoclinic one and its stabilization at room temperature is rather complicated as expected. Moreover, the nature of fixed signal, like heat changes and values of electrical current, also plays an important role in identifying the onset and end temperatures of phase transition in La2Mo2O9 crystal system. The combination of this broadband high-temperature impedance spectroscopy and differential scanning calorimetry techniques led us to find both the explanation and the relationship between electrical properties and structural changes in investigated material, especially, in the case of large particles with size of about 100–200 μm and small crystallites whose sizes do not exceed 72 nm.

Arrhenius plots of temperature dependencies of standard deviation for α and β crystal phase transition processes in La2Mo2O9 ceramic
Conclusions
For the first time, to our knowledge the aqueous tartaric acid-assisted sol–gel synthesis technique was successfully proposed for the preparation of La–Mo–O tartrate gel and La2Mo2O9 ceramic materials. The detailed TG–DSC analysis of La–Mo–O tartrate precursor clearly revealed that its thermal decomposition mainly consists of three stages during which the fully inter-oxidation of tartaric acid, partial decomposition of metal tartrates and the final burning of residue of inorganic carbon were occurred. The formation of the final ceramic with the initial composition of La2Mo2O9 cubic phase was established from the 793 to 844 K of temperature. The further DSC investigation of La2Mo2O9 ceramic revealed that heating and cooling rates play an important role in both onset and end temperatures. Besides, comparing the results obtained from the detailed investigation of La–Mo–O tartrate gel precursor and La2Mo2O9 ceramics, it is clear that by increasing the heating rate the onset temperature also increases. With the help of DSC and IS analyses, it was concluded that the phase transition from cubic to monoclinic crystal system occurs under more favorable conditions and its activation energy is slightly smaller.
References
Alekseeva OA, Antipin AM, Gagor A, Pietraszko A, Novikova NE, Sorokina NI, et al. Single-crystal structure of vanadium-doped La2Mo2O9. Crystallogr Rep. 2013;58(6):829–34.
Borah L, Paik B, Hashmi SA, Pandey A. Conductivity and electrical modulus studies of La2-x Nd (x) Mo1.7W0.3O9-delta oxygen ion conductor. Ionics. 2012;18(8):747–57.
Xia T, Li JY, Li Q, Liu XD, Meng J, Cao XQ. Synthesis, structural characterization and electrical property of new oxide ion conductors: La3MMo2O12 (M = In, Ga and Al). Solid State Ionics. 2006;177(37–38):3267–73.
Zhang YW, Yang Y, Jin S, Tian SJ, Li GB, Jia JT, et al. Sol-gel fabrication and electrical property of nanocrystalline (RE2O3)(0.08)(ZrO2)(0.92) (RE = Sc, Y) thin films. Chem Mater. 2001;13(2):372–8.
Kosacki I, Suzuki T, Petrovsky V, Anderson HU. Electrical conductivity of nanocrystalline ceria and zirconia thin films. Solid State Ionics. 2000;136:1225–33.
Zhuang Z, Wang XP, Li D, Zhang T, Fang QF. Ionic conductivity enhancement of La2Mo2−xWxO9 nanocrystalline films deposited on alumina substrates by the sol–gel method. J Am Ceram Soc. 2009;92(4):839–44.
Wang JX, Wang Q, Wang XP, Li C, Fang QF. Synthesis and characterization of fine grained high density La2Mo2O9-based oxide-ion conductors. J Mater Sci Technol. 2008;24(5):761–5.
Abdala PM, Custo GS, Lamas DG. Enhanced ionic transport in fine-grained scandia-stabilized zirconia ceramics. J Power Sources. 2010;195(11):3402–6.
Bellino MG, Lamas DG, de Reca NEW. Enhanced ionic conductivity in nanostructured, heavily doped ceria ceramics. Adv Funct Mater. 2006;16(1):107–13.
Lacorre P, Goutenoire F, Bohnke O, Retoux R, Laligant Y. Designing fast oxide-ion conductors based on La2Mo2O9. Nature. 2000;404(6780):856–8.
Liu X, Fan HQ, Li Q, Shi J. Monoclinic distortion shrinkage and enhanced properties of La2Mo2O9 oxide ion conductors synthesized by an isobaric-microwave assisted method. Ecs Solid State Lett. 2013;2(9):N27–30.
Baque L, Vega-Castillo J, Georges S, Caneiro A, Djurado E. Microstructural and electrical characterizations of tungsten-doped La2Mo2O9 prepared by spray pyrolysis. Ionics. 2013;19(12):1761–74.
Marrero-Lopez D, Pena-Martinez J, Ruiz-Morales JC, Perez-Coll D, Martin-Sedeno MC, Nunez P. Structural and electrical characterization of Nb5+ and Cr6+ substituted La2Mo2O9. Bol Soc Esp Ceram. 2008;47(4):213–8.
Georges S, Goutenoire F, Bohnke O, Steil MC, Skinner SJ, Wiemhofer HD, et al. The LAMOX family of fast oxide-ion conductors: overview and recent results. J New Mat Electr Syst. 2004;7(1):51–7.
Kolesnikova DS, Kharitonova EP, Voronkova VI. Synthesis and phase transitions of oxide-ion conducting compound La2Mo2O9 doped with alkaline metals. Crystallogr Rep. 2011;56(2):315–20.
Marrero-Lopez D, Pena-Martinez J, Perez-Coll D, Nunez P. Effects of preparation method on the microstructure and transport properties of La2Mo2O9 based materials. J Alloys Compd. 2006;422(1–2):249–57.
Zhuang Z, Wang XP, Sun AH, Li Y, Fang QF. Sol-gel synthesis and transport property of nanocrystalline La2Mo2O9 thin films. J Sol-Gel Sci Technol. 2008;48(3):315–21.
Subasri R, Matusch D, Nafe H, Aldinger F. Synthesis and characterization of (La1−xMx)(2)Mo2O9-delta; M = Ca2+, Sr2+ or Ba2+. J Eur Ceram Soc. 2004;24(1):129–37.
Wang JX, Wang XP, Liang FJ, Cheng ZJ, Fang QF. Enhancement of conductivity in La2Mo2O9 ceramics fabricated by a novel three-stage thermal processing method. Solid State Ionics. 2006;177(17–18):1437–42.
Saradha T, Muzhumathi S, Subramania A. Microwave-assisted combustion synthesis of nanocrystalline La2Mo2O9 oxide-ion conductor and its characterization. J Solid State Electr. 2008;12(2):143–8.
Cheng H, Wang HE, Li L, Lu ZG, Qian D. Facile synthesis of La2Mo2O9 nanoparticles via an EDTA complexing approach. Rare Met. 2008;27(4):340–4.
Fievet F, Lagier J, Blin B, Beaudoin B, Figlarz M. Homogeneous and heterogeneous nucleations in the polyol process for the preparation of micron and submicron size metal particles. Solid State Ionics. 1989;32–33:198–205.
Barbooti MM, Al-Sammerrai DA. Thermal decomposition of citric acid. Thermochim Acta. 1986;98:119–26.
Chattaway FD, Ray FE. II—The decomposition of tartaric acid by heat. J Chem Soc Trans. 1921;119:34–7.
Arumugam Manithiram PNK, Sundaram SK, Chan S-W. Front Matter. Developments in Solid Oxide Fuel Cells and Lithium Ion Batteries: Wiley, 2006; p. i–viii.
Kezionis A, Kazlauskas S, Petrulionis D, Orliukas AF. Broadband method for the determination of small sample’s electrical and dielectric properties at high temperatures. IEEE Trans Microw Theory. 2014;62(10):2456–61.
Kazlauskas S, Kezionis A, Kazakevicius E, Orliukas AF. Charge carrier relaxation and phase transition in scandium stabilized zirconia ceramics. Electrochim Acta. 2014;134:176–81.
Cruywagen JJ, Heyns JBB, Rohwer EA. Molybdenum(Vi) complex-formation. 4. Equilibria and thermodynamic quantities for the reactions with tartrate in 1.0-Mol Dm-3 sodium-chloride. J Chem Soc Dalton. 1990;6:1951–6.
Stankeviciute R, Zalga A. Sol-gel synthesis, crystal structure, surface morphology, and optical properties of Eu2O3-doped La2Mo3O12 ceramic. J Therm Anal Calorim. 2014;118(2):925–35.
Acknowledgements
This research was funded by a Grant (S-LZ-17-7) from the Research Council of Lithuania.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Žalga, A., Gaidamavičienė, G., Gricius, Ž. et al. Aqueous sol–gel synthesis, thermoanalytical study and electrical properties of La2Mo2O9. J Therm Anal Calorim 132, 1499–1511 (2018). https://doi.org/10.1007/s10973-018-7120-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-018-7120-3