
Abstract
An overview in the field of chemical kinetics on the thermal decomposition of di-tert-butyl peroxide (DTBP) has been performed in this study. Nowadays, DTBP has been a model compound for studying thermokinetics of organic peroxide and standardization of the DSC or adiabatic calorimeter. Thermal decompositions of DTBP in neat state or solution are conducted by heat flow or adiabatic calorimeters. Chemical kinetics on the thermal decomposition of DTBP obeyed n-th-order reaction and the type of Arrhenius equation. Order of reaction is first without any exception. DTBP in alkyl or aromatic hydrocarbon solvent behaves with excellent precision in activation energy with an averaged value of 157.0 (±4.1) and 159.7(±3.9) kJ mol−1 determined by DSC and adiabatic calorimeters, respectively. Frequency factors A (in s−1) in the form of logA are determined to be 15.8 (±1.1) and 16.3(±0.5) by DSC and adiabatic calorimeters, respectively. In the neat state of DTBP, activation energy and frequency factor in logA both possess the lower value of 128.4 (±6.2) kJ mol−1 and 12.2 (±0.8) determined by DSC. In ARC, these respective parameters are determined to be 142.0 (±17.7) kJ mol−1 and 15.5 (±1.3). Arrhenius parameters acquired from published literature with regard to the kinetics and mechanism on thermal decomposition of DTBP are summarized and discussed.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Organic peroxide is featured in possession of a weakly peroxy (–O–O–) bond in the molecule. Organic peroxide has a general formula of R–O–O–R′ in which R and R′ can symbolize a wide range of substitution groups. All typical features of reactivity or incompatibility are ascribed to the breaking of O–O bond which can undergo homolytic decomposition accompanying heat and non-condensable gases released. Most organic peroxides are either used as a curing agent or used to initiate free radical polymerization in the petrochemical industry. Fires and explosions were the ultimate types of incidents which were caused by ill-conditioned handling of organic peroxides. Incidents of fires or explosions caused by thermal decompositions of organic peroxides have been studied extensively [1, 2]. Thermal or reactive hazards rating for organic peroxide have been thoroughly discussed in the previous study [2]. Understanding the cleavage of O–O bond is fundamental to process safety, radical chemistry, and in downstream fields of petrochemical industry. O–O bond holds a generic O–O structure associating four lone pairs, and the repulsion between these lone pairs is believed to be the labile or unstable sources. The strength of the O–O bond is of great importance in the study on the kinetics of thermal decomposition of organic peroxides, and traditionally, a value of 142.2 kJ mol−1 has been ascribed to a generic O–O bond dissociation. Bach et al. have studied the bond dissociation energies of several organic peroxides by using Gaussian-2 (G2) methodology. For commercial organic peroxides, calculations by G2 method give the bond dissociation energies at 298 K of 209.2 kJ mol−1 for HOOH, 188.3 kJ mol−1 for CH3OOH, 163.2 kJ mol−1 for CH3OOCH3, 200.8 kJ mol−1 for both HC(O)OOH and CH3C(O)OOH, 159.0 kJ mol−1 for diacetyl peroxide, and 96.2 kJ mol−1 for isopropenyl hydroperoxide [3]. A new Table 1 lists the bond dissociation energy of organic peroxides reported in the literature [3–5].
The most important characteristics of thermal hazards from the studies of calorimetry stressed the exothermic onset temperature, heat of reaction, maximum temperature, adiabatic temperature rise, maximum pressure, pressure-rising rate, maximum self-heat, and time to maximum rate. Thermodynamic and kinetic data of reaction enthalpy, activation energy, pre-exponential factor, and reaction order are decisive roles for corroborating the hazard potential of unstable organic peroxides. Di-tert-butyl peroxide (DTBP) is an organic peroxide widely used as an initiator for various polymerization processes, a source for t-butoxy radical, a linking agent or hardener for unsaturated polymers, and an additive for combustion used in reforming plants. In the field of calorimetry and thermal analysis, DTBP has been used as a model compound for studying chemical kinetics under thermal decomposition and conventionally been acted as a standardization reference for a calorimeter exploring thermal hazards [6–12]. It is a member of dialkyl peroxide. Being the relatively stable peroxides, dialkyl peroxides attracted least attentions than other organic peroxides in chemical industry. But perhaps so, dialkyl peroxides have the well-behaved characterization of thermolysis or photolysis, and they have been regarded as interesting species in organic mechanism and radical chemistry [8]. DTBP acts an additive which has its thermal and oxidative stability in diesel fuels at certain temperatures, and sometimes, DTBP is recommended as an improver for diesel ignition.
Until now, the most cited work on the thermal decomposition of DTBP was performed by Shaw and Pritchard [9]. Known amounts of DTBP and carbon dioxide which could hold the pressure of 30–300 mmHg and 0–15 atm were respectively injected into stainless steel vessel. DTBP decomposed in the temperature range 90–130 °C, and at small percentage of conversions, the rate of formation of acetone measured by GC can be used as rate of decomposition of the DTBP [9]. An Arrhenius plot with first-order relation was shown; besides, the rate constants did not depend on the pressure of CO2. Activation energy and frequency factor in the form of logA (A in s−1) on the thermal decomposition of DTBP at high pressure were determined to be 158.2 ± 0.2 kJ mol−1 and 15.8 ± 0.2, respectively [9]. In an attempt to establish the standard reference materials for studying the kinetics of thermal decomposition by calorimetry, six compounds were proposed by Blaine [10]. Log A distributed from 13 to 16.5 and activation energy varied widely from 122 to 164.5 kJ mol−1, respectively [10]. Blaine claimed that the Arrhenius parameters on the thermal decomposition of DTBP reported by Shaw were the widespread values in gas phase. In a recent study on searching for kinetic reference materials for calorimetry, a table of reported kinetic parameters for the thermal decomposition on DTBP in either neat, gas or solution states was summarized by Blaine and ASTM E 2781 [10, 11]. Kersten et al. [12] also reported the Arrhenius parameters on the thermal runaway of thermal decomposition using 15 mass% DTBP determined by diverse adiabatic calorimeters. Table 2 lists the Arrhenius parameters on the thermal decompositions of DTBP and DTBP solvent reviewed by previous literature [9, 10, 12]. However, since there was significant discrepancy among these frequency factor and activation energy, no microscopic pinpoint was exaggerated to release these differences. By the practices of ASTM E698-16 and E2781-11 [11, 13], the systematic errors within laboratory and between laboratory should be respectively <4.1 and 8.4 %. Obviously, the bias of these activation energy and frequency factor in the review literature is too large to be acceptable or convincible so far. For declaring the moderate or gigantic differences in activation energy and frequency, more intensive and extensive studies are ardently desired.
For deeply investigating the dynamic behavior, thermal decomposition caught the most attentions to the chemical kinetics and unimolecular elementary reaction, which can be dissected by RRKM (Rice–Ramsperger–Kassel–Marcus) theory or ab initio calculation microscopically. In order to delicately investigate and compare the inconsistence, neat DTBP and solvated DTBP are conducted by calorimeters. In addition Arrhenius parameters in this study and published in literature are summarized and compared. Differences in activation energy and frequency factor are illustrated by decomposition pathways in gaseous DTBP, neat DTBP, and solvated DTBP. Assistance in pinpointing the linkage between macroscopic kinetics with microscopic dynamics is needed. Quantum statistical theory using Gaussian-3 (G3) software package, ab initio calculation, and RRKM theory on the explanations in exit channel of t-butoxy radical is helpful in assessing the physical scales of activation energy and frequency factor on the thermal decomposition of DTBP. Similarities or differences of reaction pathways on gaseous DTBP, neat DTBP, and solvated DTBP are presented and discussed.
Experimental
Chemicals
Di-tert-butyl peroxide (DTBP), benzene, and toluene with purity higher than 99 % were purchased from Sigma-Aldrich or Merck Company without further purification. DTBP is stored in a refrigerator at 4 °C environment for sustaining quality and purity. Neat DTBP and 20 mass% DTBP in benzene and in toluene are used as test samples. Indium metal with purity >99.9 % supplied by Mettler Co. is used as a standard for temperature and enthalpy calibration in DSC.
Differential scanning calorimeter (DSC)
Thermal curves of neat DTBP and solvated DTBP are screened in a Mettler TA-4000 System coupled with a DSC822e measuring cell [14]. Disposable crucibles (ME-26732) which can withstand to about 100 bars are used for detecting thermal curves. Data are acquired into thermal curves and stored by a PC system for further evaluation. Scanning rate is selected to be 4 °C min−1 in programmatic ramp for the reason of sustaining better thermal equilibrium inside the crucible.
Accelerating rate calorimeter (ARC)
A microprocessor-controlled accelerating rate calorimeter (ARC), manufactured by Columbia Scientific Industries of Austin, Texas [15], is utilized in this study. The detailed performance and theory of the ARC instrument were proposed by Townsend [16]. The thermokinetic and pressure behaviors on the thermal decompositions of DTBP are investigated in different thermal inertia. Three types of spherical bombs with a volume about 10 mL made of titanium, s.s. 316 and Hastelloy C can be selected.
Methodologies for acquiring kinetic parameters
Thermal analysis
Thermal decomposition of DTBP is a modeling of thermal curve exhibited in DSC or DTA. Rate constant follows the Arrhenius type, and the decomposition is a first-order reaction. Practically, this means that the exothermic curve upon thermal decomposition of DTBP must be well behaved smoothly with no shoulders, overlapped multiple peaks, shifts in baselines, or discontinuous steps. Rate equation associated with the consumption of reactant may be modeled with a number of well-established rate equations. Because of its simplest characteristics in thermal curve, methods of the Kissinger, Borchardt & Daniels, Friedman, or Flynn/Wall/Ozawa have been applied for determining the chemical kinetics or kinetic triplet on thermal decomposition of DTBP [17–23]. These methods provide the effectual means for determining activation energy, pre-exponential factor, and order of reaction by using differential thermal or isoconversional methods. The results obtained among these approaches must be comparable to each other or in agreement with the values determined by similarly respective ASTM standard methods or practices issued by committee E [11, 13, 24, 25]. Some limitations should be avoided in applying these simplified models such as autocatalytic reaction, consecutive reaction, inhibited reaction, parallel reaction, heterogeneous reaction, phase transition, thermoset curing reaction, and crystallization reaction. The programmed temperature ramp at a rate between 1 and 10 K min−1 starts from a point at least 50 K below the first observed exothermic threshold. Sample size is kept small to minimize temperature gradients within the sample. In general, a sample mass resulting in a maximum heat generation of <8 mW is satisfactory. These methodologies assume thermal equilibrium throughout the whole test specimen.
For applying to complex reaction, autocatalytic reaction, non-thermal equilibrium, self-heating within sample, and activation energy varies with conversion, and some advanced methodologies have been recommended by the Kinetics Committee of the International Confederation for Thermal Analysis and Calorimetry (ICTAC). The recommendations provide guidance for delicate evaluation of kinetic parameters from the data detected by using thermal analysis methods such as TG, DSC, and DTA. The recommendations cover the most common kinetic (such as method of Kissinger, Friedman, and Flynn/Wall/Ozawa), model-free (such as differential isoconversional and integral isoconversional methods), and model-fitting [26, 27] methods.
Methods of thermal analysis for acquiring the Arrhenius parameters
Ι. The Kissinger method
As being the earliest and most successful kinetic equation associated with thermal analysis, the Kissinger equation was introduced in 1957 [17]. This method can be used to determine the activation energy from the plot of logarithm of the heating rate versus the inverse of temperature at the maximum reaction rate by a constant heating rate. The advantage of this method lies in that the activation energy can be evaluated without knowing the accurate reaction mechanism, by using the following equation:
where β is the heating rate and T p is the temperature at maximum reaction rate.
ΙΙ. The Borchardt & Daniels method
Kinetic parameters can also be extracted by a non-isothermal method originally developed by Borchardt and Daniels [18] coupled with the non-isothermal curve detected by DSC. This method fitted only to some reactions with the simple reaction mechanism and a distinguishable curve. Heat-releasing power of an exothermic reaction (such as thermal decomposition of DTBP) in dynamic scanning can be equated as:
This equation can also be written as
or
where ΔH is enthalpy change, dH/dt is heat releasing power, A is frequency factor, E a is activation energy, n is reaction order, and α is conversion.
A multiple linear equation written in logarithmic form of the type was used for evaluating kinetic parameters:
Multiple linear regression analysis provides the underlined kinetic data using i triples of measured values (dα/dt)i, α i, and T i directly from thermal curve. These deviations of the measured data from the least-square-fit plane permit the confidence limits for 95 % probability. Kinetic parameters that exactly follow Eqs. (4) and (5) lead to very low confidence limits of n ≤ 0.05, E a ≤ 5 kJ mol−1, and lnA ≤ 1.
ΙΙΙ. The Friedman method
Having been the alternatives to the Kissinger equation, several so-called isoconversional methods were developed. In the case of this differential isoconversional method, the assumption reflected in the Kissinger equation is further extended to the premise that the degree of conversion remained constant at the certain characteristic stage of a reaction. By this approach, the activation energy can be determined for any chosen degree of conversion, assuming that the dependence of E a versus α holds the well-behaved features. Generally, the activation energy is calculated by the slope via choosing several rates of conversions (dα/dt) versus 1/T at constant α for a set of β and taking the averaged activation energy. The Friedman method is the earliest one and the most straightforward approach [19]. A simplified Friedman equation is presented as follows:
where the dα/dt and T α are the values of rate of conversion and temperature at the specified degree of conversion and a constant heating rate.
ΙV. The Flynn–Wall–Ozawa method
The Friedman method may result in erroneous value of the activation energy. To pursue much more accurate kinetic parameters than those estimated from the Friedman method, the Flynn–Wall–Ozawa method has been developed and derived from the integral isoconversional method [20–22]. By applying the Doyle approximation [23], a simplified equation of the Flynn–Wall–Ozawa method can be written as:
where A is the frequency factor and g(α) is a constant from the integral of kinetic model. Thus, the activation energy can be estimated by Flynn–Wall–Ozawa method for the reaction with any particular degree of conversion. E a is determined by the linear dependence of logβ versus 1/T at different heating rates without the necessity of knowing reaction order. Besides, the frequency factor can be deduced from the intercept which is in conjunction with the integral conversion function g(α) known. In general, at least four different heating rates are required to perform the kinetic analysis for using Flynn–Wall–Ozawa method.
Adiabatic calorimetry and Townsend’s methodology
In addition to thermal analysis from dynamic and isothermal DSC, thermal curves can discriminate chemical kinetics in neat DTBP or various solvated DTBP. The non-isothermal approach can be performed by using an adiabatic calorimeter. A model for thermal decomposition kinetics on DTBP is proposed in the following equations. Thermal decomposition kinetics of neat DTBP or 20 mass% DTBP can be deduced by temperature versus time relation or via the self-heat rate (dT/dt). A pseudo-zero-order rate constant (k*) for the n-th-order exothermic decomposition reaction can be calculated from the observed self-heat rate (dT/dt) at corresponding temperature. The experimental measurements of self-heat rate and temperature behavior in 15 or 20 mass% can be assessed in comparison with the theoretical calculation. The self-accelerating temperature to time curve shows that the experimental data on self-heat rate are in fact bounded quite close to the proposed model with a reaction order of 1.0. The pseudo-zeroth-order rate constant (k*) equates to rate constant k in first-order reaction. Furthermore, k is proportional to self-heat rate (dT/dt) in rate equation. Therefore, the Arrhenius plot can be expressed in ln(dT/dt) versus 1/T and is expected to be a straight line. A linear fitted curve is chosen from which E a (kJ mol−1), A(s−1), and n can be explicitly determined [16]. For φ > 1 and first-order reaction, self-heat rate for obtaining Arrhenius parameters can be expressed in the following equations,
This can be related to Arrhenius equation
where dT/dt is self-heat rate, φ is thermal inertia, ΔH is enthalpy change, C p is heat capacity, dα/dt is rate of conversion, ΔT AD is adiabatic temperature rise with the φ value of 1, ΔT ad is adiabatic temperature rise with the φ value >1, A is frequency factor, E a is activation energy, n is reaction order, and α is conversion.
Search for kinetic parameters on thermal decomposition of DTBP
The thermal decompositions of DTBP identified by isothermal, non-isothermal, and adiabatic modes have been implemented in diversely international laboratories. Kinetic parameters searched from calorimetric studies were collected and summarized. Data under the concentrations of DTBP solutions distributed from 10 mass% to about 60 mass% and neat DTBP were searched. These kinetic results mainly determined by the commercial calorimeters such as DSC, TAM, ARC, RSST, C-80, PHI-TEC, and VSP2. These laboratories had chosen methodology or software package for analyzing the kinetic parameters such as the method of Kissinger (ASTM E 2890) [25] method of Flynn/Wall/Ozawa (ASTM E698) [13], method of Borchardt & Daniels (ASTM E2041) [24], model-free method or method of Friedman [19], Townsend’s theory (ASTM E1981) [16, 28], Thermal safety software (TSS) [29], and Thermokinetics software (Advanced Kinetics and Technology Solutions, AKTS) [30]. Owing to the existed large discrepancy among kinetic parameters, a detailed inspection of these data was validated to discriminate the reasonable data from the published kinetic parameters. These data were judged to be acceptable or not were based on the bias of individual parameter with an uncertainty compared with the averaged value less than that recommended by ASTM E2041 [24]. The between laboratory repeatability presented in relative standard deviation suggested by ASTM E2041 for E a, logA, and reaction order should within 9.8, 9.8, and 22.0 %, respectively.
Results and discussion
Chemical kinetics
Decomposition of DTBP in gas state
Investigation on the thermal degradation of DTBP in the capillary column of a gas chromatography was performed by Wrabetz and Woog [31]. By carrying out the quantifications on the peak area in GC, calculated data contained the first-order rate constants from 130 to 170 °C. The Arrhenius parameters of activation energy and frequency factor in logA (A in s−1) were determined to be (163.6 ± 2.1) kJ mol−1 and 16.5, respectively. A set of main pathways on the thermal decay of DTBP in the gas phase was proposed as follows: (1) DTBP → 2 (CH3)3CO (2) (CH3)3CO → (CH3)2CO + CH3 (3) 2 CH3 → C2H6. In a similar way, headspace gas chromatography was employed to study the thermal decomposition of gaseous di-tert-butyl peroxide from 130 to 160 °C [31]. Cafferata and Manzione reported a homogeneous unimolecular decomposition corresponding to the rupture of O–O bond in DTBP by following the common three-stage mechanism [32]. Activation energy and frequency factor in the form of logA (A in s−1) on the thermolysis of DTBP in gaseous phase were determined to be (157.3 ± 6.1) kJ mol−1 and 15.3 ± 0.2, respectively [32]. These data on the thermal decomposition of gaseous DTBP are presented in Table 3 [9, 31, 32].
Decomposition of DTBP in neat state
Differential scanning calorimetry
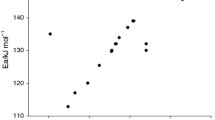
Thermal decomposition of DTBP in liquid state or in non-protonic solvent has been demonstrated to proceed a first-order reaction in which dissociation of the oxygen–oxygen bond is apparently an unimolecular and rate-determining step. Blaine had reviewed the search for the reference materials for obtaining the Arrhenius kinetics by differential and adiabatic calorimetry [10]. Though every DSC have to be calibrated, both the temperature and enthalpy change by Indium sample referred to the practices of ASTM E967-14 and ASTM E968-14 [33, 34]. Due to melting of Indium holds an endothermic behavior, researchers may always calibrate the DSC by an alternative exothermic sample. Pure DTBP and DTBP in toluene solution are the most recommended as the standard samples for verification the performance of various calorimeters. It is now a worldwide practice or a rule of thumb, and before the study on chemical kinetics of a reaction by calorimeter, chemical kinetics of DTBP should be performed in advance to assure the accuracy and precision of a calorimeter. By gathering so many data from diverse literature, both the frequency factor and activation energy distribute extremely wide. Both the kinetic parameters summarized by Shaw et al. and Blaine [9, 10], frequency factor presented in logA ranged from 12.9 to 16.63 and activation energy determined from 136 to 163 kJ mol−1, disputes left without any further explanations by microscopic theory or reaction dynamics. However, a quite smaller E a of 122.1 kJ mol−1 determined by high pressure DSC was declared by Gimzewski and Audley [35]. Duh et al. [8] reported an activation energy of 125 kJ mol−1 on the thermal decomposition in pure DTBP studied by DSC. The amazing deviations in activation energy of 80.0 and 111.4 kJ mol−1 reported respectively by Wu et al. and Liu et al., which are far away from the averaged activation energy at about 157 kJ mol−1 or O–O bond energy of 160 kJ mol−1 reassessed by Bach [3]. Enormous bias of Arrhenius parameters are suspected to be resulted from bad performance of DSC without standard or accurate calibration [36, 37]. Kinetic parameters on the thermal decompositions of neat DTBP are depicted in Table 4 [35–40]. Figure 1 shows the relation of activation energy versus logA on the thermal decomposition of neat DTBP detected by DSC.

Relation of activation energy versus logA on the thermal decomposition of neat DTBP detected by DSC
Adiabatic calorimetry
In order to acquire the complete behavior on the thermal decomposition of pure organic peroxide in an ARC bomb, mass of organic peroxide was selected about 1 to 3 g to inject into test bomb. Generally, due to the high thermal inertia about from 3 to 10, most of the kinetic parameters extracted from ARC data were significantly influenced by thermal inertia. Gimzewski and Audley reported a set of data on the thermal runaway of neat DTBP detected by ARC [35]. Activation energy was calculated to be 113.2 kJ mol−1, which was the lowest value of activation energy reported on the thermal decomposition of neat DTBP until now; besides, order and frequency factor were not described in the literature [35]. From data listed in Table 5, kinetic parameters on the thermal decompositions of neat DTBP detected by ARC exhibit lower values in comparison with the data detected in DTBP solutions [12, 35, 41–43]. Activation energy and frequency factor in the form of logA are determined to be 142.0 ± 17.7 kJ mol−1 and 15.5 ± 1.3, respectively. Figure 2 presents the relation of activation energy versus logA on the thermal decomposition of neat DTBP detected by ARC.

Relation of activation energy versus logA on the thermal decomposition of neat DTBP detected by ARC
Decomposition of DTBP in solution
DSC calorimetry
Thermal decomposition of DTBP in non-protonic solvent has been recommended as a standard sample for verification the performance of DSC and adiabatic calorimeters. 20 mass% of DTBP in toluene is the worldwide one. 20 mass% of solvated DTBP in benzene is the second alternative. We have elucidated 20 mass% DTBP solvated in toluene and benzene for evaluating the Arrhenius parameters from dynamic scanning by DSC. DTBP in each concentration had been repeated at least three times to ascertain the excellent precision and accuracy kept by DSC. Kinetic data on thermal decompositions of DTBP in solutions studied by DSC are listed in Table 6 [39, 44, 45]. Relation of activation energy versus logA on the thermal decomposition of DTBP in organic solvents detected by DSC is shown in Fig. 3. Order of reaction is first in all case. LogA (A in s−1) spreads from 14.6 to 18.1. Activation energies exhibit excellent agreement in the range of 150.8–163 kJ mol−1. Averaged activation energy and frequency factor in logA are 157.0 ± 4.1 kJ mol−1 and 15.8 ± 1.1, and these data match well with the summarized kinetic parameters reported by Shaw et al. [9] and Blaine [10].

Relation of activation energy versus logA on the thermal decomposition of DTBP in organic solvents detected by DSC
Adiabatic calorimetry
For conducting the thermal runaway of energetic compounds in the ARC bomb, mass of sample are generally limited to <3 g for avoiding the rupture of the test system. Thermal inertia is thus in the range of 3–10. For a larger phi-factor transcending >5, the corrected data will apart from the real value without any realistic meaning for runaway hazards. Even using delicate analytical equation for correcting the effects of phi-factor, data related to thermodynamics or enthalpy change such as adiabatic temperature rise shall be corrected accurately. However, data relative to chemical kinetics, for example, self-heat rate, adiabatic time to maximum rate or pressure-rising rate, cannot be recovered as precise as real runaway with the phi-factor approaching unity. Therefore, scientists consecrated to the following three approaches to liberate the problems distorted by phi-factor. First of all, using large quantity of diluted sample to occupy the volume ratio of about 80 % in ARC bomb is commonly adopted for lowering phi value and overcoming the defect of large phi-factor in adiabatic calorimeters. The second method was to use a thin cell of can shape for holding sample about 50 mL, in which cell keeping a maximum volume of 112 mL was thereafter developed to be capable of improving the phi-factor as low as 1.1. Some researchers instead have established the theoretical approach to simulate thermal runaway, especially for the well-known energetic compounds or explosives [35, 44, 46, 47]. Self-heat rate at set pressure and time to maximum rate are both crucial parameters used for the design for emergency relief system in industrial reactor in case of runaway encountered. On this base, phi-factor of 20 mass% DTBP in toluene can be <1.3 in a standard ARC bomb. Temperature and pressure data of thermal runaway at the phi-factor <1.3 shall possess excellent adiabatic condition with at most 23 % of heat sunk into bomb. Thermal runaway of solvated DTBP will thus exhibit practical behavior performed by adiabatic calorimeters.
A set of comparable activation energies from 138 to 167 kJ mol−1 with several different decomposition pathways had been reported by Iizuka and Surianarayanan [43]. In their 10 ARC tests, the phi-factor lied between 1.33 and 2.66. Kersten et al. obtained the more precise activation energy from 155.4 to 159.3 kJ mol−1 by various adiabatic calorimeters in the results of round robin tests [12]. The round robin experiments were conducted in the ARC, Phi-Tec, pressure Dewar calorimeter (Dewar), Automatic Pressure Track Accelerating Calorimeter (APTAC) and the Controlled Runaway and Vent Monitor (CRVM) with an individual phi-factor value from 1.05 to 1.47. Table 7 compiles the kinetic data from the implementation on the thermal decompositions of DTBP solutions studied by diverse adiabatic calorimeters [6, 12, 40, 41, 43, 47–49]. Relation of activation energy versus logA on the thermal decomposition of DTBP in organic solvents detected by adiabatic calorimetry is depicted in Fig. 4. Averaged activation energy and frequency factor in logA are 159.7 ± 3.9 kJ mol−1 and 16.3 ± 0.5, which are extremely close to the results obtained from gas phase by GC and DTBP solutions by DSC.

Relation of activation energy versus logA on the thermal decomposition of DTBP in organic solvents detected by adiabatic calorimetry
In some previous studies, a so-called isokinetic effect has been observed and widely reported in the literature [50–52]. The validity of the isokinetic effect or compensation effect in the field of kinetics analysis has been debated heatedly. Generally, this effect can be simplified by a linear relation between logA and activation energy E a,
In which a and b are constants. The occurrence of isokinetic effect can be caused by chemical or physical factors or can arise from the computational artifact. Some results in literature showed that the various methods for solving kinetic equation, i.e., in evaluating logA and E a, resulted in this isokinetic or compensation effect. Besides, the compensation trends have also reported by the International Congress on Thermal Analysis and Calorimetry (ICTAC) kinetics project [53]. In this study Figs. 1, 2, and 4 display explicitly the isokinetic effect kept within these three figures.
Mechanisms of thermal decomposition
From the above summarized data in previous literature, kinetic parameters relevant to the thermal decomposition on DTBP detected by various calorimeters varied significantly. To our knowledge, there is seldom interpretation on the major reasons on the differences of these frequency factors and activation energies. The interpretation of the Arrhenius parameters in relation to the reaction mechanism seemed to be a hard defiance. Traditionally, only simple mechanism consisting of three elementary reactions linked the chemical kinetics. Iizuka and Surianarayanan first broke the tie bound at the pinpoint between Arrhenius parameters and mechanism [43]. They took into account the physical processes and chemical transformations for studying the kinetics on the thermal decomposition of DTBP. By combining GC-total inorganic carbon (TIC), GC–MS, ARC, and software of BatchCAD, Arrhenius parameters on thermal decompositions of DTBP and DTBP solvent systems were studied in an extensive way to solve the existing paradox. However, chemical kinetics had not been told from these three different mechanism linked to neat DTBP, DTBP in toluene, and DTBP in benzene. In short, Iizuka and Surianarayanan draw a conclusion that the Arrhenius equation on thermal decomposition of neat DTBP and DTBP solvent is a common one. Only overall activation energy and logA (A in s−1) were reported to be 154.7 kJ mol−1 and 17.4 in their conclusion. In this study, Arrhenius parameters related to the thermal decomposition of DTBP and DTBP solvents are deduced from the previous studies as a whole. Table 8 depicts the classified Arrhenius parameters on the thermal decompositions of DTBP gathered by various methodologies until now [6, 31, 32, 43]. Mechanisms for explaining the thermal decomposition of neat or solvated DTBP are depicted as following:
Decomposition mechanism of DTBP in gaseous state
T-butoxy radical [(CH3)3CO] can be generated at initial stage by the thermal-assisted homolysis on DTBP molecule. The fates of t-butoxy radical depend on the molecules encountered or radicals itself. In DTBP with purity >99 % in liquid state or gaseous states, DTBP was immune to the radical attack [9]. Cleavage or elimination of t-butoxy radical via tertiary carbon and β C–C scission is the only path to penetrate the reaction scheme. The t-butoxy radical can proceed an elimination by giving up a methyl in breaking tertiary C–C bond. Acetone and ethane are therefore predicted to be dominated in the final products. This conventional mechanism for explaining the thermal decomposition of DTBP in gaseous state is presented as following:
Decomposition mechanism of neat DTBP
The above mechanism is identical to the decomposition mechanism of gaseous DTBP proposed in early literature [31, 32]. To explain the products detected in GC-TIC, GC–MS and to link chemical kinetics, Iizuka and Surianarayanan proposed an exquisite mechanism for thermal decomposition of neat DTBP as follows [43].
In the earlier study, Bell et al. [54] proposed a similar mechanism on the thermal decomposition of neat DTBP. They discovered that the decomposition of DTBP in various solvents has been shown to proceed by a first-order process in which scission of the O–O bond is apparently an unimolecular and rate-determining step. The principal products are t-butyl alcohol, acetone, and methane. In an unlike manner, then pure DTBP in the liquid phase undergoes decomposition, isobutylene oxide is a major reaction product, and the rate of peroxide breakdown is accelerated. This alteration in the reaction is attributed to attack on the peroxide by its own decomposition fragments when hydrocarbon solvent is removed without a hydrogen donating source.
Mechanism for thermal or photochemical decomposition in neat DTBP was proposed:
Decomposition mechanism of DTBP in organic solvents
From Tables 6 and 7, thermal decompositions of DTBP were conducted in benzene, toluene, pentadecane, and silicone oil. T-butoxy radical can be generated at initial stage by the thermal-assisted homolysis on DTBP molecule. The exit channels of t-butoxy radical depend on the molecules or radicals collided. Beta C–C scission resulting in an elimination of methyl radical via tertiary carbon and or hydrogen abstraction can be the possible way to go through the reaction scheme. Both the acetone and t-butyl alcohol are therefore predicted to be paramount in the final products [43]. Mechanism for explaining the thermal decomposition of DTBP in solvated state is presented as following:
Decomposition of DTBP in toluene proposed by Iizuka and Surianarayanan [43]
Decomposition of DTBP in toluene proposed by Aldeeb et al. [6]
Decomposition of DTBP in benzene or benzene/biphenyl by Iizuka and Surianarayanan [43]
Theoretical calculations
Arrhenius parameters in relation to transition state theory
An unimolecular O–O bond dissociation was assumed in theoretical treatment of dissociative phenomenon [3]. For an elementary reaction with single molecularity, reaction order is unity, E a and A can be expressed from transition state theory. In a simplified treatment, E a = ΔH 0≠ + RT and logA = (1/2.303)[ΔS 0≠/R + ln(ek B T/h)]. For example, by using the data of enthalpy of activation (ΔH 0≠) and entropy of activation (ΔS 0≠) [3], T is assumed to be the exothermic onset temperature at about 120 °C, Arrhenius parameters on the thermal decomposition of RO-OR or (CH3)3CO–OC(CH3)3 (DTBP) can be calculated to be E a = 150.6 + 3.3 = 153.9 kJ mol−1 and logA (A in s−1) = 3.1 + 12.9 = 16.0. Transition state theory and parameters in Table 9 present the calculated activation energy and frequency factor, which agree excellently with the results derived from experiments in Table 8.
Exit channels for t-butoxy radical
Thermal decomposition and product reaction pathways
In previous study, Aldeeb et al. [6] applied the computational quantum chemistry for simulating the pathways under the thermal decomposition of DTBP in toluene. A set of computational software in quantum chemistry composed of semiempirical (AM1), Hartree–Fock (HF), density functional theory (DFT) B3LYP, and Gaussian-2 were implemented in the calculation. They concluded that formation of t-butoxy radical will be the rate-determining step, and then t-butoxy radical will participate in the cleavage by β C–C bond scission or react with toluene to propagate the chain reaction. Due to the lower Gibbs free energy in the exit channel of β C–C bond scission, the ratio of acetone production was expected to be higher than the ratio of the H abstraction with solvents. Being the latest results on theoretical study focused at the thermal decompositions of DTBP, Sebbar et al. [55] proposed the detailed simulation on plausible decomposition paths by using density function theory [DFT; B3LYP/6-311 g(d,p)], and composite ab initio G3MP2B3 calculations. They found no saddle point at transition state (transition state 1, TS1) for the DTBP dissociation. At the first elementary reaction, DTBP dissociates homolytically into two t-butoxy radicals by the collision of thermal motions. About 10 initial reaction pathways were simulated and calculated theoretically. Three lower barrier reaction pathways (from about 184 to 222 kJ mol−1) contain two t-butoxy radicals (pathway 1), t-butyl-hydroperoxide plus isobutene (pathway 2), and t-butoxy radical plus a hydroxyl t-butyl alkyl radical (pathway 3). DTBP homolyzed into two t-butoxy radicals holding the intrinsically lowest barrier energy of 184 kJ mol−1. However, competing channel of H abstraction from solvent by t-butoxy radical was not included in this study. Besides, reaction products from pathway 2 and pathway 3 are never detected by any experiment until now due to the higher barrier than that of homolysis. In short, the first intermediate on the thermal decomposition is t-butoxy radical. The most crucial step is the following cleavage of t-butoxy radical by β C–C scission or via the hydrogen abstraction from organic solvent. This step devotes the fates of t-butoxy radical related to resulting products and macroscopic Arrhenius parameters.
Only β C–C scission or mixed with H abstraction
Thermal unimolecular decomposition of t-butoxy radical, usually as a model radical for studying chemistry of free radical reaction or atmospheric intermediates, possessed the most representative β C–C scission in various alkoxy radicals. It is also recognized to be a prototype for the bond dissociation from β C–C scission that occurred at larger alkoxy radical. Fittschen et al. [56] had studied and disclosed the temperature dependence of the unimolecular decomposition of t-butoxy radical by the laser photolysis/laser-induced fluorescence (LIF) technique. This is the most key step which correlates and results in the reaction order, activation energy and frequency factor in the thermal decomposition of dialkyl peroxides and t-butyl peroxyacetate. By using both the ab initio calculation and density function theory (DFT) in computing the rate constant for the β C–C bond scission of t-butoxy radical, a common pre-exponential factor for β C–C bond scission rate constants of all alkoxy radicals of log A = 14 was suggested [56]. The ab initio study on the potential energy surface predicts the limiting rate constant from RRKM (Rice–Ransperger–Kassel–Marcus) theory. The simulation excluded the exit channels of β C–H bond dissociation and 1,3-hydrogen transfer isomerizations which were both energetically unfavorable. E 0 (kJ mol−1), E a (kJ mol−1), ΔH 0≠(kJ mol−1), ΔS 0≠(J mol−1 K−1), and logA(s−1) for the β C–C bond scission of t-butoxy radical were calculated to be 56.7, 61.2, 58.3, 14.0, and 14.0, respectively [56]. Buback et al. [57] announced another similar work on the rate coefficients of β C–C bond scission in tertiary alkoxy radical, R(CH3)2CO has been verified by using DFT calculation in conjunction with transition state theory. Alkyl group strongly influences the magnitude of β C–C bond scission rate in several orders.
In order to prove the applicability of the DFT-derived rate constant, Buback et al. [57] inspected the product distributions from the thermal decomposition of t-butyl peroxyacetate by using chromatography. The branch ratio of alcohol-to-ketone product has been identified after the complete decomposition of t-butyl peroxyacetate in n-heptane. By measuring the ratio of alcohol-to-ketone product, which is proportional to the rate ratio of channel ΙΙΙ (alcohol or “ol” formation by hydrogen abstraction) to channel ΙΙ (ketone or “on” formation by β C–C scission), alcohol or ketone concentrations depended on temperature can be analyzed by GC. Arrhenius plots determined by the gas chromatography detected alcohol-to-ketone product ratio results in the straight lines with the slope being defined as the difference in activation energies, ΔE a (ol/on), of the intramolecular hydrogen abstraction and β scission. Thus, a fatal quantity presented the difference in activation energy, and ΔE a (ol/on) was termed by Buback [57]. ΔE a (ol/on) deduced from decomposition studies was determined to be −36.4 kJ mol−1. The energy difference performed by UB3LYP/6-31G(d,p) was calculated to be ΔE a (ol/on) = −35.5 kJ mol−1.
The β-scission of the t-butoxy radical to generate an acetone molecule and a methyl radical has been known worldwide. However, in solution states, the hydrogen abstraction of t-butoxy radical competes with β-scission itself in the existence of various solvents or substrates. The microscopic kinetics and branching ratios on both the hydrogen abstraction and β scission of the butoxy radical had ever been an interesting and attractive research topic. A tight transition state model on the evaluation of the Arrhenius parameters for the dissipation reaction for the alkoxy radical has been proposed by Choo and Benson [58]. An estimated logA (A in s−1) = 14.1 and E a = 60.0 kJ mol−1 of the t-butoxy radical decomposition reactions coincide with the rate constants detected in different environments [58]. On the analogy of results, the phenomena of hydrogen abstraction and/or β scission have been studied using laser flash photolysis by Tsentalovish et al. [59], using chromatography by Soon and Choo [60], evaluating delayed radical using time-resolved electron spin resonance by Weber et al. [61]. Table 10 lists the Arrhenius parameters and rate constants at 298 K for the elementary reactions of the t-butoxy radical reported [55–61]. From the kinetic parameters in Table 10, both the hydrogen abstraction and β scission are confirmed to possess a strong and a weak solvent dependence, respectively.
Effects of aromatic and aliphatic hydrocarbon solvents
The dissipating pathways of hydrogen abstraction by the t-butoxy radical with solvent system of benzene or toluene will involved the resonant benzyl radical. As shown in Table 10 the hydrogen abstraction rates of substituted toluene or benzoyl structure and the rate constant (k H, 298) decrease to the order of 104. For the effects of substituted toluenes upon the reactivity of hydrogen abstractions with t-butoxy radicals, Kennedy and Ingold had studied the relative rates of hydrogen abstractions from ten substituted toluenes by t-butoxy radicals in CCl4 at 40 °C [62]. The rate of hydrogen abstraction can be frequently correlated by means of the Hammett equation [62]. They concluded that the structure of XC6H6C+H2·H−:OBu made a cardinal contribution to the transition state then favored the hydrogen abstraction reaction. The reactivity of hydrogen abstraction of t-butoxy radical with toluenes containing the substituents of p-CH3, m-CH3, none, p-Cl, m-Cl, m-NO2, and p-NO2 had the relative ratios of 1.47, 1.07, 1.00, 0.89, 0.59, 0.29 and 0.25 in reaction rates, respectively. Their results implied that in no case the reactivity was significantly enhanced by the stability of the resultant benzyl radical with a resonant or a mesomeric structure. This fact reveals that the hydrogen abstraction between t-butoxy radical and substituted toluenes, the inductive characteristics of the substituents, play the much more important role than the stabilizing ability on the benzyl radical in the reaction pathway of hydrogen abstraction. One of the decisive reasons is that the E a and k H is related to the exit channels of the t-butoxy radical by bimolecular collision. The k H and activation energy of substituted toluenes are approximately 1.0 log units lower and 10 kJ mol−1 higher than these parameters determined for aliphatic hydrocarbon solvents without the transient benzyl structure. The increase in E a can be attributed to a difference in the configuration of transition states between the substituted toluenes or t-butoxy radicals. Having the benzyl group with higher steric hindrance, this stiffened complex shall lead to a reduction in the reactivity owing to the loss of entropy of activation resulted from an increase in the barrier to molecular rotation.
Conclusions
Chemical kinetics on the thermal decomposition of gaseous DTBP, neat DTBP, and DTBP solution are overviewed in this study. DTBP in alkyl or aromatic hydrocarbon solvent behaves with excellent precision in activation energy with an averaged value of 157.0 (±4.1) and 159.7(±3.9) kJ mol−1 determined by DSC and adiabatic calorimeters, respectively. Frequency factors A (in s−1) in the form of logA are determined to be 15.8 (±1.1) and 16.3(±0.5) by DSC and adiabatic calorimeters, respectively. In the neat state of DTBP, activation energy and frequency factor in logA both possess the lower value of 128.4 (±6.2) kJ mol−1 and 12.2 (±0.8) determined by DSC, respectively. In ARC, these respective parameters are determined to be 142.0 (±17.7) kJ mol−1 and 15.5 (±1.3). The paradoxical differences on the activation energy and frequency factor in the neat DTBP in previous literature are summarized and validated. To increase the sample mass of neat DTBP injected into the ARC bomb for reducing the phi-factor <1.5 is still a challenge. More theoretical and experimental efforts are suggested to focus at the thermal decomposition of neat DTBP by DSC. In the near future, some more dainty methodology or theoretical approaches for solving the dispute are needed. In particular, some astonishing large deviations in the frequency factor from calorimetric data between 1011 and 1017, which fact was already known but never resolved by any calorimetry or theoretical approaches. Calorimetric determinations on the Arrhenius parameters with superior accuracy and precision will still be a hard fight. Though keeping the lower activation energy in either the hydrogen abstraction or β C–C bond scission governed by t-butoxy radical, the activation energy on the overall thermal decomposition is measured to be about 158.0 (±4.0) kJ mol−1. Microscopic dynamics which can declare the linkage between the reaction mechanism and the complicated decomposition of DTBP are feverishly expectant.
Abbreviations
- A :
-
Frequency factor [s−1 M1−n]
- a,b,c :
-
Constant [dimensionless]
- C p :
-
Heat capacity at constant pressure [kJ kg−1 K−1]
- C pb :
-
Heat capacity of test bomb at constant pressure [kJ kg−1 K−1]
- C ps :
-
Heat capacity of reactant at constant pressure [kJ kg−1 K−1]
- D :
-
Bond dissociation energy [kJ mol−1]
- E a :
-
Activation energy [kJ mol−1]
- E 0 :
-
Activation energy in transition state theory [kJ mol−1]
- ΔE a :
-
Activation energy difference between Hydrogen abstraction and β C–C scission [kJ mol−1]
- e :
-
Natural exponential [dimensionless]
- g(α):
-
A constant from the integral of kinetic model [dimensionless]
- ΔH :
-
Heat of reaction [kJ kg−1]
- ΔH 0≠ :
-
Enthalpy of activation
- h :
-
Planck constant [6.6262 × 10−34 J s]
- k :
-
Rate constant [s−1 M1−n]
- k 2 :
-
Rate constant of recombination [s−1 M1−n]
- k B :
-
Boltzmann constant [1.38 × 10−23 J K−1]
- k D :
-
Rate constant of hydrogen abstraction from DTBP [s−1 M1−n]
- k e :
-
Rate constant of ethane formation [s−1 M1−n]
- k H :
-
Rate constant of hydrogen abstraction from solvent [s−1 M1−n]
- k β :
-
Rate constant of β scission [s−1]
- m b :
-
Mass of test bomb used in adiabatic calorimeter [kg]
- m s :
-
Mass of reactant [kg]
- n :
-
Order of reaction [dimensionless]
- R :
-
Ideal gas constant [8.314 J g−1 K−1]
- ΔS 0≠ :
-
Entropy of activation
- T 0 :
-
Onset temperature of exothermic reaction [K]
- T max :
-
Maximum temperature of exothermic reaction [K]
- T p :
-
Temperature of maximum reaction rate [K]
- T α :
-
Temperature at a specified degree of conversion [K]
- ΔT AD :
-
Adiabatic temperature rise with the φ value of 1 [°C or K]
- ΔT ad :
-
Adiabatic temperature rise with the φ value >1 [°C or K]
- X i, Y i, Z i :
-
Constant of linear regression [dimensionless]
- dH dt −1 :
-
Heat-releasing power [Ws−1]
- dT dt −1 :
-
Self-heat rate [°C min−1]
- α :
-
Degree of conversion [dimensionless]
- β :
-
Heating rate of calorimeter [°C min−1]
- ϕ :
-
Thermal inertia [dimensionless]
- dαdt −1 :
-
Rate of conversion [dimensionless]
- β :
-
β C–C scission
- H:
-
Hydrogen abstraction
References
Ho TC, Duh YS, Chen JR. Case studies of incidents in runaway reactions and emergency relief. Process Saf Prog. 1998;17:259–62.
Duh YS, Wu XH, Kao CS. Hazard ratings for organic peroxides. Process Saf Prog. 2008;27:89–99.
Bach RD, Ayala PY, Schlegel HB. A reassessment of the bond dissociation energies of peroxides. An ab Initio study. J Am Chem Soc. 1996;118:12758–65.
Benson SW, Shaw R. Thermochemistry of organic peroxides, hydroperoxides, polyoxides, and their radicals. In: Swern D, editor. organic peroxides. New York: Wiely; 1970. p. 251–306.
Matsugo S, Saito I. Dialkyl peroxides. In: Ando W, editor. Organic peroxides. New York: Wiely; 1992. p. 157–94.
Aldeeb AA, Rogers WJ, Mannan MS. Theoretical and experimental methods for the evaluation of reactive chemical hazards. Process Saf Environ Prot. 2002;80:141–9.
Duh YS, Wang WF, Kao CS. Novel validation on pressure as a determination of onset point for exothermic decomposition of DTBP. J Therm Anal Calorim. 2014;116:1233–9.
Duh YS, Yo JM, Lee WL, Kao CS, Hsu JM. Thermal decompositions of dialkyl peroxides studied by DSC. J Therm Anal Calorim. 2014;118:339–47.
Shaw DH, Pritchard HO. Thermal decomposition of di-tert-butyl peroxide at high pressure. Can J Chem. 1968;46:2722–4.
Blaine R. The search for kinetic reference materials for adiabatic and differential calorimetry. J Therm Anal Calorim. 2011;106:25–31.
ASTM E2781-11 Standard practice for evaluation of methods for determination of kinetic parameters by thermal analysis.
Kersten RJA, Boers MN, Stork MM, Visser C. Results of a Round-Robin with di-tertbutyl peroxide in various adiabatic equipment for assessment of runaway reaction hazards. J Loss Prev Proc Ind. 2005;18:145–51.
ASTM E698-16 Standard test method for arrhenius kinetic constants for thermally unstable materials using differential scanning calorimetry and the Flynn/Wall/Ozawa Method.
TA4000 operation instructions, Mettler Company; 1993.
Varions ARC. Manual. Austin: C.S.I.
Twonsend DI, Tou JC. Thermal hazard evaluation by an accelerating rate calorimeter. Thermochim Acta. 1980;37:1–30.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29:109–17.
Borchardt HJ, Daniels F. The application of differential thermal analysis to the study of reaction kinetics. J Am Chem Soc. 1957;79:41–6.
Friedman HL. Kinetics of thermal degradation of char-forming plastics from thermo-gravimetry. Application to a phenolic plastic. J Poly Sci C. 1965;50:183–95.
Ozawa T. A new method analyzing thermogravimetric data. Bull Chem Soc Jpn. 1965;38:1881–6.
Ozawa T. Thermal analysis-review and prospect. Thermochim Acta. 2000;355:35–42.
Flynn JH, Wall LA. General treatment of the thermogravimetry of polymers. J Res Natl Bur Stand Sect A. 1966;70:487–523.
Doyle C. Estimating isothermal life from thermogravimetric data. J Appl Polym Sci. 1962;6:639–42.
ASTM E2041-13, Standard test method for estimating kinetic parameters by differential scanning calorimeter using the Borchardt and Daniels method.
ASTM E2890-12, Standard test method for estimating kinetic parameters for thermally unstable materials by differential scanning calorimeter using the Kissinger method.
Vyazovkin S, Chrissafis K, Griado JM, Perez-Maqueda LA, Popescu C, Sbirrazzuoli N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis. Thermochim Acta. 2011;520:1–19.
Vyazovkin S, Burnham AK, Di Lorenzo ML, Koga N, Pijolat M, Roduit B, Sbirrazzuoli N, Sunol JJ. ICTAC kinetics committee recommendations for collecting experimental thermal analysis data for kinetic computations. Thermochim Acta. 2014;590:1–23.
ASTM E1981-12 Standard guide for assessing thermal stability of materials by methods of accelerating rate calorimetry.
Thermal safety software. Cheminform St. Petersburg Ltd., St. Petersburg.
Advanced kinetics and technology AG.
Wrabetz K, Woog J. Investigation of the thermal degradation of di-tert-butyl peroxide in the capillary column of a gas chromatograph. Fres Zeit Anal Chem. 1987;329:487–9.
Cafferata LFR, Manzione CJ. Kinetics and mechanism of gas-phase thermolysis using headspace-gas chromatographic analysis. J Chromatogr Sci. 2001;39:45–8.
ASTM E967-14 Standard test method for temperature calibration of differential scanning calorimeters and differential thermal analyzers.
ASTM E968-14 Standard practice for heat flow calibration of differential scanning calorimeters.
Gimzewski E, Audley G. Thermal hazards: calculating adiabatic behavior from DSC data. Thermochim Acta. 1993;214:129–40.
Wu SH, Chou HC, Pan RN, Huang YH, Hirng JJ, Chi JH, Shu CM. Thermal hazard analyses of organic and inorganic peroxides by calorimetric approaches. J Therm Anal Calorim. 2012;109:355–64.
Liu SH, Lin CP, Shu CM. Thermokinetic parameters and thermal hazard evaluation for three organic peroxides by DSC and TAM ΙΙΙ. J Them Anal Calorim. 2011;106:165–72.
MacNeil DD, Trussler S, Fortier H, Dahn JR. A novel hermetic differential scanning calorimeter sample crucible. Thermochim Acta. 2002;386:153–60.
Hou HY, Liao TS, Duh YS, Shu CM. Thermal hazards studies for dicumyl peroxide by DSC and TAM. J Therm Anal Calorim. 2006;83:167–71.
Chu YC, Chen JR, Tseng JM, Tsai LC, Shu CM. Evaluation of runaway thermal reactions of di-tert-butyl peroxide employing calorimetric approaches. J Them Anal Calorim. 2011;106:165–72.
Tou J, Whiting L. The thermokinetic performance of an accelerating rate calorimeter. Thermochim Acta. 1981;48:21–42.
Duh YS, Kao CS, Hwang HH, Lee WWL. Thermal decomposition kinetics of cumene hydroperoxide. Process Saf Environ Prot. 1998;76:271–6.
Iizuka Y, Surianarayanan M. Comprehensive kinetic model for adiabatic decomposition of di-tert-butyl peroxide using BatchCAD. Ind Eng Chem Res. 2003;42:2987–95.
Wang WY, Duh YS, Shu CM. Evaluation of adiabatic runaway reaction and vent sizing for emergency relief from DSC. J Therm Anal Calorim. 2006;85:225–34.
Torfs JCM, Deij L, Dorrepaal AJ, Heijens JC. Determination of Arrhenius kinetic constants by differential scanning calorimetry. Anal Chem. 1984;56:2863–7.
Sanchirico R. Adiabatic behavior of thermal unstable compounds evaluated by means of dynamic scanning calorimetric (DSC) techniques. AIChE J. 2013;59:3806–15.
Kossoy AA, Singh J, Koludarova EY. Mathematical methods for application of experimental adiabatic data—an update and extension. J Loss Prev Proc Ind. 2015;33:88–100.
Leung J, Fauske H, Fisher H. Thermal runaway reactions in a low thermal inertia apparatus. Thermochim Acta. 1986;104:13–29.
Iwata Y. Thermal decomposition behavior of di-tert-butyl peroxide measured by differential adiabatic calorimeter. Chem Eng Trans. 2013;31:1–6.
Agrawal RK. On the compensation effect. J Them Anal Calorim. 1986;31:73–86.
Mianowski A, Bigda R. The Kissinger law and isokinetic effect Part Ι. Most common solutions of thermokineic equations. J Them Anal Calorim. 2003;74:953–73.
Mianowski A, Bigda R. The Kissinger law and isokinetic effect Part ΙΙ. Experimental analysis. J Them Anal Calorim. 2004;75:355–72.
Brown ME, Galwey AK. The significance of “Compensation effects” appearing in data published in “Computational aspects of kinetic analysis”: ICTAC project, 2000. Thermochim Acta. 2002;387:173–83.
Bell ER, Rust FF, Vaughan WE. Decompositions of dialkyl peroxides. IV. Decomposition of pure liquid peroxide. J Am Chem Soc. 1950;72:337–8.
Sebbar N, Bozzelli JW, Bockhorn H. Kinetic study of di-tert-butyl peroxide: thermal decomposition and product reaction pathways. Int J Chem Kinet. 2015;47:133–61.
Fittschen C, Hippler H, Viskolcz B. The β C–C bond scission in alkoxy radicals: thermal unimolecular decomposition of t-butoxy radicals. Phys Chem Chem Phys. 2000;2:1677–83.
Buback M, Kling M, Schmatz S. Decomposition of tertiary alkoxy radicals. Z Phys Chem. 2005;219:1205–22.
Choo KY, Benson SW. Arrhenius parameters for the alkoxy radical decomposition reactions. Int J Chem Kinet. 1981;13:833–44.
Tsentalovich YP, Kulik LV, Gritsan NP, Yurkovskaya AV. Solvent effect on the rate of β-scission of the tert-butoxyl radical. J Phys Chem. 1998;102:7975–80.
Weber M, Fischer H. Absolute rate constants for the β-scission and hydrogen abstraction reactions of the tert-butoxyl radical and for several radical rearrangements: evaluating delayed radical formations by time-resolved electron spin resonance. J Am Chem Soc. 1999;121:7381–8.
Song SA, Choo KY. Study of the kinetics and mechanisms of alkoxy radical reactions in the gas phase (Ι). Arrhenius parameters for t-butoxy radical reactions with isobutene and cyclohexane. Bull Kore Chem Soc. 1984;5:16–21.
Kennedy BR, Ingold KU. Reactions of alkoxy radicals Ι. Hydrogen atom abstraction from substituted toluenes. Can J Chem. 1966;44:2381–5.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Duh, YS., Kao, CS. & Lee, WL.W. Chemical kinetics on thermal decompositions of di-tert-butyl peroxide studied by calorimetry. J Therm Anal Calorim 127, 1071–1087 (2017). https://doi.org/10.1007/s10973-016-5859-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5859-y