
Abstract
New aminoguanidinium metal oxalate complexes with the formulae (AmgH)2[Cu(C2O4)2] and (AmgH)2[Cd(C2O4)2(H2O)2] have been synthesized and characterized. The copper compound loses aminoguanidine exothermically at 240 °C in DTA, whereas aminoguanidine is lost endothermically at 200 °C in cadmium. Both cases decompose exothermically via their respective metal oxalate intermediates to give metal oxide as the end product. It was also observed that hydrazine is lost exothermically in the hydrazinium copper oxalate hydrate. The single-crystal X-ray diffraction study of the copper complex revealed that aminoguanidinium ions are not involved in coordination but act as charge-compensating cations. It is interesting to note that both oxalates act as bidentate chelating ligands. One oxalate bridges the neighbouring copper atom through its carbonyl oxygen with a bond length of 2.561 Å to form square pyramidal geometry around the copper atom.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Oxalic acid is a dicarboxylic acid that has acquired great importance because its versatile coordination modes can provide novel complexes with properties such as magnetism and luminescence [1, 2]. Structurally, the oxalate anion is well known for its chelating coordination mode and symmetric bis-chelating bridging mode. The prevalence of this linking mode provides a degree of predictability with regard to structural motifs and architectures found in the oxalate coordination network [3–7]. Consequently, many 1D, 2D and 3D networks have been characterized. The most common metal oxalate structure is the 2D honeycomb structure. A large number of studies have been performed on transition metal oxalates with different organic bases such as ammonia [8], ethylenediamine [9], hydrazine [10], piperazine [11, 12], benzylamine, propylenediamine [13] and guanidine [14]. Among these, hydrazine metal carboxylates have been intensively studied, and oxalate complexes are widely used as precursors for the synthesis of spinel oxides [15, 16]. To the best of our knowledge, there is no report on guanylhydrazine (aminoguanidine) metal complexes of carboxylates in general and oxalate in particular.
Our research group has long been involved in studies on the preparation and thermal behaviour of hydrazinium salts with different aliphatic [17, 18] and aromatic [19, 20] carboxylic acids, as well as their corresponding metal (mostly transition and lanthanide metals) complexes. We are specifically interested in studying the coordination ability and thermal stability of hydrazine in the presence of different types of carboxylic (aliphatic, aromatic and hetroaromatic) acids and their corresponding metals. We recently reported the thermal properties of different simple hydrazinium salts and metal hydrazine complexes of heterocyclic carboxylic acids such as 2-pyrazinecarboxylic acid, 2,3-pyrazinedicarboxylic acid [21], 4,5-imidazoledicarboxylic acid [22] and 3,5-pyrazoledicarboxylic acid [23]. Previous reports have described the preparation and thermal reactivity of hydrazinium 2,n-pyridinedicarboxylates (n = 3, 4, 5, and 6) [24] and metal dipicolinate complexes of hydrazine [25]. Fortified by laboratory results on the thermal behaviour of other metal carboxylate and/or metal hydrazine carboxylate systems [26–30], we extended our study to the coordination ability and thermal reactivity of aminoguanidine (substituted hydrazine) in the presence of carboxylic acids with divalent transition metals. Oxalic acid, which has two carboxylic groups and four active coordination sites, was selected for its potential in chelating coordination and symmetric bis-chelating bridging coordination to central metal cations. Although the coordination behaviour of aminoguanidine is not exactly the same as that of hydrazine, aminoguanidine can act as a monodentate or bidentate chelating [31] ligand towards metal ions. It is well known that hydrazinium ions can behave as charge-compensating cations in various metal carboxylate systems. We sought to determine whether aminoguanidinium can also function as a charge-compensating counter cation in metal carboxylate systems in general and the metal oxalate system in particular. This is the first attempt to synthesize aminoguanidinium metal oxalates and compare the coordinating ability and thermal properties of their metal complexes with those of hydrazinium metal oxalates.
Experimental
Materials and methods
All reagents used for synthesis were obtained commercially and used as received. Elemental analysis of C, H and N was performed on an Elementar Vario EL III analyser. The FTIR spectra were recorded on an AVATAR 320 FTIR spectrometer using KBr pellets in the range of 4000–400 cm−1. X-ray powder diffraction was recorded on a Rigaku diffractometer with copper radiation (λ = 0.154 06 nm) at 40 kV and 40 mA. Simultaneous TG–DTA curves were recorded on a Perkin Elmer Pyris Diamond instrument at a heating rate of 5 °C min−1 under air. Absorption spectral measurements were carried out using a JASCO V-530 UV–visible spectrophotometer in the range of 200–800 nm. Quartz cuvettes with 1 cm path lengths were used to record the absorption spectra. Hydrazine content was estimated by titrating against standard KIO3 under Andrews’ condition [32]. Metal contents were determined by EDTA titrations [33].
Preparation of (AmgH)2[Cu(C2O4)2]
The compound was prepared by adding a mixture containing 20 mL aqueous solution of copper acetate (0.05 g; 0.25 mmol) and aminoguanidine bicarbonate (0.136 g; 1 mmol) in a 1:4 molar ratio to an aqueous solution (20 mL) of oxalic acid (0.126 g; 1 mmol) neutralized with sodium carbonate (0.106 g; 1 mmol). The turbidity was resolved with addition of a few drops of 2 N acetic acid. The resulting clear blue solution was concentrated to one-third of its volume in a water bath and maintained at room temperature for crystallization. The blue needle-like crystals, suitable for single-crystal X-ray studies, formed after four days were separated, washed with dry ethanol and air-dried.
Preparation of (AmgH)2[Cd(C2O4)2(H2O)2]
An aqueous solution (20 mL) of oxalic acid (0.126 g; 1 mmol) was added to the aqueous solution (20 mL) containing cadmium nitrate hexahydrate (0.077 g; 0.25 mmol) and aminoguanidine bicarbonate (0.136 g; 1 mmol). To this solution, a few drops of 2 N nitric acid were added to prevent turbidity. The resulting clear solution was concentrated to half of its volume and maintained at room temperature for crystallization. After 2 days, the white crystalline solid (found to be guanylhydrazido-oxalic acid) that formed was filtered off. Following evaporation, the filtrate was a polycrystalline solid that was separated as in the previous preparation.
Single-crystal structure determination
The X-ray diffraction data of (AmgH)2[Cu(C2O4)2] was collected using a single crystal on a Bruker Smart Apex CCD diffractometer with graphite-monochromated MoKα (λ = 0.71073 Å) radiation. Crystals were mounted in Fluorlube oil on a Mitegen mount or glass loop and placed in a cold N2 stream 203(1) K for data collection. The structure was determined using direct methods and refined by full-matrix least squares on F2 using the SHELXL-97 software package [34]. Each structure was solved via direct methods that determine the positions of non-hydrogen atoms. Typically, all non-hydrogen atoms were refined anisotropically.
Final difference Fourier syntheses showed only chemically insignificant electron density (with the largest difference peaks close to metal atoms). Inspection of F o versus F c values and trends based upon sinθ, Miller index, or parity group failed to reveal any systematic error in the data. Detailed crystallographic data are listed in Table 1.
Results and discussion
Synthesis
Gajapathy et al. [10] have prepared hydrazinium copper oxalate monohydrate by reacting copper nitrate trihydrate with hydrazinium oxalate in a ratio 1:4. Aminoguanidinium copper oxalate was prepared using the respective metal acetate hexahydrate and oxalic acid dihydrate with aminoguanidine bicarbonate in the presence of acetic acid and sodium carbonate. The pH of the final solution was 4–5. With copper acetate as the starting material, single crystals resulted, whereas the other copper salts resulted in polycrystalline compounds of the same composition. When reactions were carried out in 1:2:2 and 1:4:4 (metal:ligand:base) mole ratios, stable copper oxalate precipitated. The cadmium complex reaction carried out with a 1:4:4 (metal nitrate:acid:base) mole ratio surprisingly yielded two products. The detailed synthetic procedure is shown in Scheme 1. The preparation of complexes using other transition metals (e.g., Co, Ni and Zn) was unsuccessful due to the formation of respective metal oxalates as insoluble products. The results of chemical analysis of the complexes are given in Table 2 and are best fit with the proposed compositions.

Reaction scheme showing the preparation of aminoguanidinium metal (M) oxalate (M = Cu or Cd)
Structural description of (AmgH)2[Cu(C2O4)2]
The structural analysis of aminoguanidinium copper oxalate (Fig. 1) revealed that the crystal contains discrete (AmgH)+ cations and complex [Cu(C2O4)2]2− anions. The coordination stereochemistry of the CuO5 chromosphere is square pyramidal. Four oxygen atoms from two nearly planar bidentate chelate oxalates occupy the basal coordination positions. A neighbouring carbonyl oxygen (O8) from an adjacent unit facilitates the apical metal coordination to produce a one-dimensional staircase-like chain. This chain runs parallel to the c axis, as shown in Fig. 2. The apical atom, O8, has an elongated Cu–O bond of 2.561 Å, while the copper-oxygen distances (Table 3) in the square base range from 1.9383(17) to 1.9465(17) Å. The bite angles of the bidentate oxalate anion [O1–Cu–O2 and O5–Cu–O6] are 85.55 and 85.92°. These values are normal and comparable with the value of the N2H5 + complex [10]. The aminoguanidinium cations are almost planar and have normal bond lengths, as expected [35, 36]. The packing pattern shows strong interlocking between complex anions and cationic sub-lattices through a multidirectional hydrogen bonding (Table 4) network, as shown in Fig. 3.

Asymmetric Cu complex unit with a thermal ellipsoid shown at 60 % probability

1D staircase structure of the Cu complex

2D hydrogen bonding pattern down the a axis
Infrared spectra
For copper and cadmium complexes, the asymmetric and symmetric stretching frequency of carboxylate ions are seen at 1620 and 1607 cm−1 and 1408 and 1384 cm−1, respectively. The average separation Δν (νasym – νsym) of 212 and 223 cm−1 indicates monodentate coordination for the carboxylate ion, as shown in Fig. 4. The iminidine peak and N–N stretching vibrations around 1680 cm−1 (Fig. 4a inset) and 1100 cm−1, respectively, indicate the presence of aminoguanidine in the crystal. For the copper complex (Fig. 4a), the peak around 3355 cm−1 is attributed to N–H stretching of aminoguanidine. For the cadmium complex (Fig. 4b), the sharp peak around 3410 cm−1 is assigned to O–H stretching of coordinated water molecules.

Infrared spectra of a (AmgH)2[Cu(C2O4)2] and b (AmgH)2[Cd(C2O4)2(H2O)2]. Inset shows the expansion range (1850–1500 cm−1) of (b)
Electronic spectra
The electronic spectrum of the copper complex is shown in Fig. 5. The complex displays a lower energy band at 671 nm (14,900 cm−1), which corresponds to the transition from the B 1 (\( d_{{\text {x}^{2} - \text{y}^{2} }} \)) ground state to the excited A 1 (\( d_{{\text {z}^{2} }} \)), B 2 (d xy) and E (d xz, d yz) states. This d–d transition at 671 nm indicates that the copper has a square pyramidal geometry [37]. The higher energy intense transitions at 249 and 224 nm are due to intraligand π → π* and n → π* charge transfer transitions (ILCT), respectively.

Electronic spectra of (AmgH)2[Cu(C2O4)2] ILCT (left) and d–d transition (right)
Thermal study
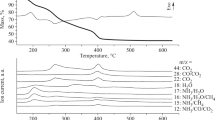
The copper complex shows two clear decomposition steps in TG (Fig. 6a). In the first step, aminoguanidine and one of the oxalate ligands decompose exothermically at 240 °C to give copper oxalate compound. The copper oxalate further decomposes with the two exothermic peaks (380 and 540 °C) to give copper oxide as the final residue. It was anticipated that the two endothermic peaks indicate different chemical changes. The first peak at around 380 °C may corresponds to the elimination of CO (carbon monoxide) to become CuCO3, while the second peak at around 540 °C corresponds to the elimination of CO2 (carbon dioxide) to become CuO as the final product. Albeit, the DTA shows two distinct exothermic peaks, the TG shows continuous decomposition which restricts our effort to calculate the exact mass loss for the corresponding steps.

Simultaneous TG–DTA of a (AmgH)2[Cu(C2O4)2] and b (AmgH)2[Cd(C2O4)2(H2O)2]
TG of cadmium compound shows (Fig. 6b) two distinct steps for the successive loss of two water molecules in accordance with two endothermic dehydrations at 110 and 150 °C (Table 5). The anhydrous compound was unstable (we could not able to isolate), which then endothermically decomposes (200 °C) to yield a cadmium oxalate compound. Like copper oxalate, the cadmium oxalate also decomposes exothermically at 500 °C to give CdO as the final residue. The exothermic shoulder peak observed around 260 °C may be attributed to the elimination of CO to form CdCO3 intermediate, which continuously decomposes to form CdO as the end residue. The only difference between the two reactions is that the anhydrous copper compound decomposes endothermically, whereas the decomposition product of anhydrous cadmium compound decomposes exothermically, probably due to their structural differences. The complete thermal decomposition details are given in Table 5.
The IR spectra of the decomposition products, copper (Fig. 7a) and cadmium oxalate (Fig. 7b), register a characteristic strong band in the region 1620 cm−1 and 1310 cm−1, which were ascribed to the asymmetric and symmetric stretching vibrations of oxalate (O–C=O) groups [38], respectively. Further, the strong and sharp peak observed around 770–790 cm−1 is assigned to the effect of in-plane deformation of O–C=O as well as the presence of a metal–oxygen coordination bond [38]. Further, the XRD patterns of copper oxalate (Fig. 7c) and cadmium oxalate (Fig. 7d) were well matched with that of their JCPDS file No 21-297, and 140712, respectively, which confirms the formation of respective products [39, 40].

Infrared spectra of the decomposition products a copper oxalate and b cadmium oxalate. The powder XRD pattern of c copper oxalate d cadmium oxalate
The IR spectra of the final decomposition products such as CuO and CdO are given in Fig. 8. A strong IR characteristic absorption band (Fig. 8a) observed at around 535 cm−1 was assigned to the Cu–O stretching vibration [41]. The IR spectrum of CdO (Fig. 8b) register a characteristic band at around 470 cm−1 with poorly resolved shoulder at 550 cm−1, which revealed the formation of CdO [42] powder. Further, it can be clearly seen that the characteristic vibration of cubic CdO occurs in the band ranging from 410 to 1100 cm−1 with the clear peak at 1050 cm−1 indicating the formation of cubic CdO [43].

IR spectra of a CuO and b CdO (the spectral region range from 1200 to 400 cm−1 is shown here for the clarity). The powder XRD patterns of c CuO and d CdO
Further, the final thermal decomposition products of metal complexes were authentically confirmed as CuO and CdO by using their powder XRD data. The XRD peaks of CuO and CdO (Fig. 8) are very sharp and intense, indicating their pure and crystalline nature. According to the JCPDS data (JCPDS No. 45-0937), the diffraction peaks of the CuO (Fig. 8c) can be indexed to be monoclinic CuO phase, which is in good agreement with the results reported in the literatures [39, 41]. The phase of the CdO (Fig. 8d) obtained from thermal decomposition of cadmium complex can be indexed to be cubic CdO structure (JCPDS No. 05-0640) [42, 43] and no characteristic peaks of impurities were detected.
Our efforts to separate the decomposition intermediates such as anhydrous cadmium complex, (AmgH)2[Cd(C2O4)2], and the anticipated respective metal carbonates in their pure forms were ineffective due to their continuous decompositions, as evident in the TG curve. The likely intermediates were assigned using observed TG mass losses, which are in agreement with calculated mass losses.
X-ray powder pattern
The X-ray powder patterns of these compounds were recorded and are shown in Fig. 9. The compounds show decidedly different diffraction patterns, indicating their structural dissimilarity. This is expected since the compositions themselves are different. The sharp powder XRD lines indicate that these compounds are highly crystalline in nature.

Powder XRD patterns of copper and cadmium complexes
Conclusions
New aminoguanidinium metal oxalate complexes with the formulae (AmgH)2[Cu(C2O4)2] and (AmgH)2[Cd(C2O4)2(H2O)2] have been synthesized and characterized. In DTA, the copper compound loses aminoguanidine exothermically at 240 °C. In cadmium, aminoguanidine is lost endothermically at 200 °C. Both cases decompose exothermically via their respective metal oxalate intermediates to give metal oxide as the end product. In hydrazinium copper oxalate hydrate, the hydrazine is lost exothermically. Hence, these complexes may be used as a precursor to fine metal oxide particles due to the low temperature of decomposition [44].
Single-crystal X-ray diffraction study of the copper complex revealed that the aminoguanidinium ions are not involved in coordination but act as charge-compensating cations. It is interesting to note that both oxalates act as bidentate-chelating ligands. One oxalate bridges the neighbouring copper atom through its carbonyl oxygen with a bond length of 2.561 Å to form a square pyramidal geometry around the copper atom.
Supplementary material
Crystallographic data for structural analysis reported in this paper have been deposited in the Cambridge Crystallographic Data Center with CCDC number 1032144. Copies of this information may be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (fax: +44-1223-336033; e-mail:deposit@ccdc.cam.ac.uk).
References
Armentano D, Munno G-De, Lloret F, Palii AV, Julve M. Novel chiral three-dimensional iron(III) compound exhibiting magnetic ordering at Tc = 40 K. Inorg Chem. 2002;41:2007–13.
Song J-L, Mao J-G. New types of blue, red or near IR luminescent phosphonate-decorated lanthanide oxalates. Chem Eur J. 2005;11:1417–24.
Salami TO, Marouchkin K, Zavilliji PY, Oliver SRJ. Three low-dimensional tin oxalate and tin phosphate materials: BING-4, -7, and -8. Chem Mater. 2002;14:4851–7.
Vaithiyanathan R, Natarajan S, Rao CNR. Synthesis of a hierarchy of zinc oxalate structures from amine oxalates. J Chem Soc Dalton Trans. 2001;5:699–706.
Vaithianathan R, Natarajan S, Cheetham AK, Rao CNR. New open-framework zinc oxalates synthesized in the presence of structure-directing organic amines. Chem Mater. 1999;11:3636–42.
Natarajan S, Vaithianathan R, Rao CNR, Ayyappan S, Cheetham AK. Layered tin(II) oxalates possessing large apertures. Chem Mater. 1999;11:1633–9.
Ayyappan S, Cheetham AK, Natarajan S, Rao CNR. Tin(II) oxalates synthesized in the presence of structure-directing organic amines: members of a potentially vast class of new open-framework and related materials. Chem Mater. 1998;10:3746–55.
Novosad J, Messimeri AC, Papadimitriou CD, Veltsistas PG, Woollins JD. Copper(II) oxalate and oxamate complexes. Transit Metal Chem. 2000;25:664–9.
Cai J, Zhang Y, Hu X, Feng X. Tris(ethylenediamine-N, N′)cobalt(III) oxalate perchlorate dehydrate. Acta Crystallogr. 2000;C56:661–3.
Gajapathy D, Govindarajan S, Patil KC, Manohar H. Synthesis, characterisation and thermal properties of hydrazinium metal oxalate hydrates: crystal and molecular structure of hydrazinium copper oxalate monohydrate. Polyhedron. 1983;2:865–73.
Keene TD, Ogilvie HR, Hursthouse MB, Price DJ. One-dimensional magnetism in new, layered structures: piperazine-linked copper and nickel oxalate chains. Eur J Inorg Chem. 2004;5:1007–13.
Keene TD, Hursthouse MB, Price DJ. Stabilization of discrete [Cu(C2O4)2(H2O)2]2— dianions in the solid state by an extensive hydrogen bonded network. Z Anorg Allg Chem. 2004;630:350–2.
Bloomquist DR, Hansey JJ, Landee CP, Willett RD, Buder R. Structure and magnetic properties of two two-dimensional copper oxalates: bis(benzylammonium) bis(oxalato)cuprate(II) and propylenediammonium bis(oxalato)cuprate(II). Inorg Chem. 1981;20:3308–14.
Golic L, Bulc N. Structure of guanidinium tris(oxalato)chromate(III) monohydrate. Acta Crystallogr. 1988;C44:2065–8.
Gajapathy D, Patil KC. Mixed metal oxalate hydrazinates as compound precursors to spinel ferrites. Mater Chem Phys. 1983;9:423–38.
Patil KC, Gajapathy D, Pai Verneker VR. Low temperature cobaltite formation using mixed metal oxalate hydrazinate precursor. J Mater Sci Lett. 1983;2:272–4.
Yasodhai S, Govindarajan S. Preparation and thermal behaviour of some hydrazinium dicarboxylates. Thermochim Acta. 1999;338:113–23.
Yasodhai S, Govindarajan S. Hydrazinium oxydiacetates and oxydiacetate dianion complexes of some divalent metals with hydrazine. J Therm Anal Calorim. 2000;62:737–45.
Kuppusamy K, Sivasankar BN, Govindarajan S. Preparation, characterisation and thermal properties of some new hydrazinium carboxylates. Thermochim Acta. 1995;259:251–62.
Vairam S, Govindarajan S. New hydrazinium salts of benzene tricarboxylic and tetracarboxylic acids—preparation and their thermal studies. Thermochim Acta. 2004;414:263–70.
Premkumar T, Govindarajan S. Transition metal complexes of pyrazinecarboxylic acids with neutral hydrazine as a ligand. J Therm Anal Calorim. 2005;79:115–21.
Premkumar T, Govindarajan S. The chemistry of hydrazine derivatives—thermal behavior and characterisation of hydrazinium salts and metal hydrazine complexes of 4, 5-imidazoledicarboxylic acid. Thermochim Acta. 2002;386:35–42.
Premkumar T, Govindarajan S. Divalent transition metal complexes of 3, 5-pyrazoledicarboxylate. J Therm Anal Calorim. 2006;84:395–9.
Saravanan K, Govindarajan S. Preparation and thermal reactivity of hydrazinium 2, n-pyridinedicarboxylates (n = 3, 4, 5 and 6). J Therm Anal Calorim. 2003;73:951–9.
Saravanan K, Govindarajan S. Dipicolinate complexes of main group metals with hydrazinium cation. Proc Indian Acad Sci (Chem Sci). 2005;114:25–36.
Premkumar T, Govindarajan S. Thermoanalytical and spectral properties of new rare-earth metal 2-pyrazinecarboxylate hydrates. J Therm Anal Calorim. 2005;79:685–9.
Premkumar T, Govindarajan S, Xie R, Pan W-P. Preparation and thermal behaviour of transition metal complexes of 4, 5-imidazoledicarboxylic acid. J Therm Anal Calorim. 2003;74:325–33.
Yasodhai S, Govindarajan S. Hexavalent uranium dicarboxylates with hydrazine. Preparation, characterization and thermal studies. J Therm Anal Calorim. 2002;67:679–88.
Yasodhai S, Govindarajan S. Hydrazinium oxydiacetates and oxydiacetate dianion complexes of some divalent metals with hydrazine. J Therm Anal Calorim. 2000;62:737–45.
Premkumar T, Govindarajan S. Thermoanalytical and spectroscopic studies on hydrazinium lighter lanthanide complexes of 2-pyrazinecarboxylic acid. J Therm Anal Calorim. 2010;100:725–32.
Aitken DJ, Albinati A, Gautier A, Husson HP, Morgant G, Nguyen-Huy D, Jiri K, Lemoine P, Ongeri S, Rizzato S, Viossat B. Platinum(II) and palladium(II) complexes with N-aminoguanidine. Eur J Inorg Chem. 2007;21:3327–34.
Jeffery GH, Bassett J, Mendham J, Denny RC “Vogel’s Textbook of Quantitative Chemical Analysis”, 5th Ed.,1986.
Vogel AI. A text book of quantitative inorganic analysis. 4th ed. London: Longman; 1986.
Sheldrick GM, SHELXTL, version 6.10; Bruker analytical X-ray systems, Madison, WI, 2001.
Ross CR II, Bauer MR, Nielson RM, Abraham SC. Aminoguanidinium(1+) pentafluorozirconate: multiple redetermination and comparisons. Acta Crystallogr. 2002;B58:841–8.
Romanenko GV, Savelyeva ZA, Podberezskaya NV, Alekseev VI, Larionov SV. Aminoguanidinium cations and the square-bipyramidal hexachlorocuprate(II) anion in the (CH8N4 )2[CuCl6 ] crystal structure. J Struct Chem. 1994;35:317–23.
Roy S, Mitra P, Patra AK. Cu(II) complexes with square pyramidal (N2S)CuCl2 chromophore: Jahn-Teller distortion and subsequent effect on spectral and structural properties. Inorg Chim Acta. 2011;370:247–53.
Dalal PV. Nucleation controlled growth of cadmium oxalate crystals in agar gel and their characterization. Indian J Mater Sci. 2013;682950:1–5.
Jia Z, Yue L, Zheng Y, Xu Z. The convenient preparation of porous CuO via copper oxalate precursor. Mater Res Bull. 2008;43:2434–40.
Janet CM, Viswanath RP. Large scale synthesis of CdS nanorods and its utilization in photo-catalytic H2 production. Nanotech. 2006;17:5271–7.
El-Trass A, ElShamy H, El-Mehasseb I, El-Kemary M. CuO nanoparticles: synthesis, characterization, optical properties and interaction with amino acids. Appl Surf Sci. 2012;258:2997–3001.
Aldwayyan AS, Al-Jekhedab FM, Al-Noaimi M, Hammouti B, Hadda TB, Suleiman M, Warad I. Synthesis and characterization of CdO nanoparticles starting from organometalic dmphen-CdI2 complex. Int J Electrochem Sci. 2013;8:10506–14.
Askarinejad A, Morsali A. Syntheses and characterization of CdCO3 and CdO nanoparticles by using a sonochemical method. Mater Lett. 2008;62:478–82.
Selvakumar R, Geib SJ, Premkumar T, Govindarajan S. Synthesis, structure and thermal properties of a new 1D magnesium sulfoacetate coordination polymer- A precursor for MgO. J Therm Anal Calorim. 2015;121:943–9.
Acknowledgements
This paper was supported by the Faculty Research Fund, Sungkyunkwan University, 2013.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Selvakumar, R., Geib, S.J., Premkumar, T. et al. Synthesis, spectroscopic, thermal and XRD studies of aminoguanidinium copper and cadmium oxalates. J Therm Anal Calorim 124, 375–385 (2016). https://doi.org/10.1007/s10973-015-5136-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-5136-5