
Abstract
Metal–organic frameworks (MOFs) have promising practical applications in gas storage, separation and fine purification of substances, and also in catalysis. These MOFs are crystalline compounds consisting of metal ions coordinated by bridging organic ligands with the formation of porous structures. We study the kinetic stability of two inclusion compounds on the base of a such framework: [Zn2(bdc)2(dabco)]·nL (bdc2− = terephthalate, dabco = 1,4-diazabicyclo[2.2.2]octane, L = cyclohexane and benzene). The cyclohexane compound is more stable than the benzene compound, although the kinetic diameters of the guest molecules are almost equal. So as the centrosymmetrical cyclohexane molecule can easily transform the chair conformation to the bath conformation, it can have the influence on the steric hindrance (as well as on the activation barrier) for the guest molecules removal. Therefore, the entropy contribution is as favorable factor, as the energetic one in the kinetic stability of the supramolecular compounds.
Similar content being viewed by others

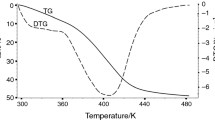
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Metal–organic coordination polymer frameworks (MOFs) are crystalline compounds consisting of metal ions coordinated by bridging organic ligands, which form multi-dimensional porous structure. MOFs with rigid and open skeleton have received intense attention for their potential applications in catalysis, gas storage, molecular recognition, high-capacity adsorbents, nonlinear optics, magnetics and biomedical imaging [1–9].
The standard process of the MOF production begins from the synthesis of the inclusion compound in the solution. The molecules of used organic solvent, such as dimethylformamide, tetrahydrofuran, dioxane, benzene, are caught in the channels and holes of the metal–organic polymer structure.
These primary included guest molecules are excluded further by the evacuation or by the heating; this process is called the framework activation. Such polymeric matrix with the empty pores, and without the support of the included guest molecules, can be thermodynamically or kinetically unstable and collapse after the guest molecules removal [10]. The stability both of the inclusion compound and of the guest-free framework can be connected with the linker ligand size or can depend on the structure of the overall coordination polyhedron [11, 12]. Therefore the quantity estimation of the stability both of the primary inclusion compound and of the guest-free host matrix (the activated framework) is important for the evaluation and the comparison properties of MOFs in the series.
Experimental
Thermal analysis
TG measurements were carried out on a Netzsch thermal analyzer TG 209 F1. The experiments were performed under a helium flow (60 cm3 min−1) at heating rates of 5, 10, 20 and 40 K min−1. The sample mass was kept ≈ 5.0 mg.
Compounds syntheses
The synthesis and the structures of the guest-free framework compound [Zn2(bdc)2(dabco)] were described earlier [13].
[Zn2(bdc)2(dabco)]·4C6H6
The crystals of the framework compound [Zn2(bdc)2(dabco)] (100 mg) were mixed together with benzene (200 cm3) and leave at room temperature for 48 h. The colorless crystals of [Zn2(bdc)2(dabco)]·4C6H6 were separated by filtration and dried on air.
[Zn2(bdc)2(dabco)]·3.5C6H12
The crystals of the framework compound [Zn2(bdc)2(dabco)] (100 mg) were mixed together with cyclohexane (200 cm3) and leave at room temperature for 48 h. The colorless crystals of [Zn2(bdc)2(dabco)]·3.5C6H12 were separated by filtration and dried on air.
In the guest-free framework, the wide open channels (7.5 × 7.5 Å2) running along the c axis are interlinked by smaller windows (ca. 4 Å) along the a and b axes, which are still large enough for the passage of small gas molecules. The guest-accessible volume is estimated to be 62 %.
The interesting features of these compound series are the structural change between the guest-free framework and the inclusion compounds. Contrary to common host–guest materials, the empty framework shrinks upon guest inclusion and expands upon guest release. The porous metal–organic framework [Zn2(bdc)2(dabco)] responds to the guest inclusion in different ways depending on the nature of the guest. The guest-free framework has the specific volume 1,148 Å3 per Zn2–unit; it shrinks upon benzene guest inclusion to 1,114 Å3, and it shrinks upon dimethylformamide guest inclusion to 1,092 Å3 [13]. So the empty flexible framework adapts to guest entering: It is forced “to outline” the guest molecules.
Kinetic analysis under non-isothermal conditions
Thermogravimetric data were processed with the computer program Netzsch Thermokinetics 2 (version 2004.05) [14, 15]. A special program module, “Model-free,” based on well-known studies [16–25], allows one to process multiple thermogravimetric curves obtained with different heating rates and calculate the activation energy without preliminary information about the kinetic topochemical equation. The Friedman method was used to calculate the activation energies for each experimental point of fractional conversion (in the range 0.005 < α < 0.995).
If the activation energy is variable in compliance with the Friedman method, therefore the decomposition process is the multistage reaction.
We further used the same set of experimental data to search for the corresponding topochemical equation (the selection was made from 16 equations: chemical reaction at the interface, nucleation and diffusion). This calculation was made by the improved differential procedure of Borchardt–Daniels [14, 26] within the multiple linear regression approach. It is very important that the range for the degree of conversion (α) for this calculation be chosen based on the relative constancy of the calculated kinetic parameters from the Friedman analysis.
The F test [14] was used to search for the best kinetic description and for statistical control of the obtained equation. It tests the residual variance of individual models against one another and answers the question of whether the models differ significantly (statistically) or not. If F exp(1) ≈ F exp(2) for two equations, there is no reason to assume the first model is better at characterizing the experiment. The statistical quantile F crit is obtained for a level of significance of 0.05.
If the calculation results in two or three kinetic equations with close values in their correlation coefficients and on the F test, but with noticeably different values in kinetics parameters, it is most correct to choose the equation with activation energy values closest to the data from the “Model-free” module program. Discrimination between the two steps is very relative in this search for topochemical equations, but it helps to find the most reliable ones. The special program of nonlinear regression is useful in searching for a full set of kinetic parameters for multistage processes. The closest fit between the activation energies from the ‘‘Model-free’’ analysis and the nonlinear regression calculation is important from a physicochemical point of view. Therefore, the authors of the computer program used recommend fixing E values (obtained by linear regression and congruent with E from the ‘‘Model-free’’ analysis) in calculations with this program.
The random error in the activation energy values for such a reversible decomposition reaction is usually about 10 % in these experiments, which we took into consideration. The computer program Netzsch Thermokinetics 2 enables estimation of the contribution of each stage (as Δm portion) after this nonlinear regression calculation.
Well-known recommendations for performing kinetic computations on thermal analysis data [27, 28] were used; new studies on non-isothermal kinetics were taken into account [29, 30].
There were several important assumptions and limitations. The kinetic equations to calculate the kinetic parameters are topochemical ones, and the calculated parameters (E and A) are formal and conventional from the standpoint of the classical chemistry of solids.
However, the general trend in the variation of these values within a specially selected series of compounds (either isostructural or genetically related) is very important because the expected disorder in the reaction zones can be identical for them; all other errors will be minimized and smoothed in such a comparison. The best series are coordination compounds with volatile ligands (with one central atom and different ligands or with different central atoms and the same ligand) [31–37]. We studied and compared the inclusion compounds with the same host matrix and the different guest molecules.
Results and discussion
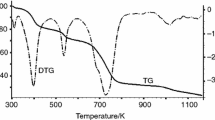
The inclusion compound with benzene is more stable than the inclusion compound with cyclohexane (Fig. 1).

Thermal decomposition of inclusion compounds: [Zn2(bdc)2(dabco)]·3.5C6H6 (1) and [Zn2(bdc)2(dabco)]·3.5C6H12(2); helium flow 60 cm3 min−1; heating rate 10 K min−1
The inclusion compound [Zn2(bdc)2(dabco)]·3.5C6H12
The decomposition at 300–460 K was chosen for the kinetic study; it corresponds to cyclohexane removal: \( \left[ {{\text{Zn}}_{2} \left( {\text{bdc}} \right)_{2} \left( {\text{dabco}} \right)} \right] \cdot 3.5{\text{C}}_{6} {\text{H}}_{12} \to \left[ {{\text{Zn}}_{2} \left( {\text{bdc}} \right)_{2} \left( {\text{dabco}} \right)} \right] + 3.5{\text{C}}_{6} {\text{H}}_{12} \uparrow \) (Fig. 2). Calculated mass loss is 34.0 %; found mass loss is 33.5 %.

Thermal decomposition of [Zn2(bdc)2(dabco)]·3.5C6H12; the heating rates were 5(1), 10(2) and 20(3) K min−1
‘‘Model-free’’ data are given in Fig. 3. The activation energy can be considered as variable in compliance with the Friedman method; therefore, the decomposition process is the multistage reaction. The best description of the process is a two-stage process (A → B → C) with a Avrami–Erofeev equation (An) and n-order equation (Fn) (Table 1, Fig. 4).

Friedman analysis of [Zn2(bdc)2(dabco)]·3.5C6H12 thermal decomposition: activation energies depending on the degree of conversion α. Perpendicular lines SD of calculation

Data processing for [Zn2(bdc)2(dabco)]·3.5C6H12 thermal decomposition. TG curve fitting of nonlinear regression, simulated with two consecutive reactions (equations An and Fn). The points are the experimental data; the lines are the calculated data. The heating rates were 5 (1), 10 (2) and 20 (3) K min−1
The most probable estimate is two consecutive reactions (Fig. 4):
Corr. coeff. = 0.999567. The time dependencies of the yield for each reactant in the decomposition are shown in Fig. 5. The mentioned mass loss step Δm = 33.5 % is related to 3.5 cyclohexane molecules removal. The used computer program enables estimation of the contribution of each stage (as Δm portion) after the nonlinear regression calculation. If 3.5 molecules removal is related to 100 % of this step of decomposition, the first stage (A → B) corresponds to 72.8 % and the second stage (B → C) corresponds to 27.2 % of this decomposition step. The approximate composition of the intermediate phase (B) is [Zn2(bdc)2(dabco)]·C6H12; it is kinetically hindered metastable phase.

Thermal decomposition of [Zn2(bdc)2(dabco)]·3.5C6H12. Time dependence of the yield of each reactant in the decomposition. The calculation corresponds to two-stage consecutive reactions (A → B → C) in Fig. 4; heating rate 20 K min−1
The inclusion compound [Zn2(bdc)2(dabco)]·4 C6H6
The calculated mass loss for [Zn2(bdc)2(dabco)]·4C6H6 (Fig. 6) compound is 35.3 %, found mass loss is 35.5 %. The decomposition of [Zn2(bdc)2(dabco)]·3.5C6H6 phase (at 340–500 K) was chosen for the kinetic study; it corresponds to full benzene removal.

Thermal decomposition of [Zn2(bdc)2(dabco)]·4C6H6; the heating rates were 5 (1), 10 (2), 20 (3) and 40 (4) K min−1
The Friedman analysis was carried out in much the same way as for the first compound. The activation energy can be considered as variable one, therefore the decomposition process is the multistage reaction. The best description of the process is a two-stage process (A → B → C) with a separate Avrami–Erofeev equation (An) for both stages (Table 2, Fig. 7).
Corr. coeff. = 0.999525. The time dependencies of the yield for each reactant in the decomposition are shown in Fig. 8. The mass loss for this step is ≈31.0 %; it is related to 3.5 benzene molecules removal. The used computer program enables estimation of the contribution of each stage (as Δm portion) after the nonlinear regression calculation. If 3.5 molecules removal is related to 100 % of this step of decomposition, the first stage (A → B) corresponds to 32.6 %, the second stage (B → C) corresponds to 67.4 % of this decomposition step. The approximate composition of the intermediate phase (B) is [Zn2(bdc)2(dabco)]·2.36C6H6; it is kinetically hindered metastable phase.

Data processing for [Zn2(bdc)2(dabco)]·3.5C6H6 thermal decomposition. TG curve fitting of nonlinear regression, simulated with two consecutive reactions (equations An and An). The points are the experimental data; the lines are the calculated data. The heating rates were 5 (1), 10 (2), 20 (3) and 40 (4) K min−1

Thermal decomposition of [Zn2(bdc)2(dabco)]·3.5C6H12. Time dependence of the yield of each reactant in the decomposition. The calculation corresponds to two-stage consecutive reactions (A → B → C) in Fig. 4; heating rate 20 K min−1
The existence of [Zn2(bdc)2(dabco)]·3.5C6H6 and [Zn2(bdc)2(dabco)]·2.36C6H6 phases of variable composition was confirmed before: they were obtained by the saturation of the framework [Zn2(bdc)2(dabco)] with the benzene vapor at different temperatures and benzene pressure [38]. It turns out now that [Zn2(bdc)2(dabco)]·2.36C6H6 compound is the kinetically hindered phase during the thermal decomposition of the inclusion compound [Zn2(bdc)2(dabco)]·3.5C6H6. It is worth to note that the crystal structure was earlier studied really for the composition ≈[Zn2(bdc)2(dabco)]·2C6H6 [13].
Two stages in the decomposition process, the appearance of the kinetically hindered phases and the change of the topochemical equations are alike for both compounds decomposition; it is due to the similarity in the framework structure transformation.
It may be understandable if the transition from the shrunk structure of the inclusion compounds to the expanded one takes place just during the kinetically hindered phases’ formation; the residual guest molecules remove from the already expanded structure.
Conclusions
The inclusion compound with cyclohexane is considerably more stable than the benzene compound in compliance both with the temperature intervals of the decomposition (Fig. 1) and with the kinetic parameter values (E a = 131/111 kJ mol−1, E b = 99/48 kJ mol−1).
The guest boiling temperatures (80.8 and 80.1 °C), evaporation heats (29.97 and 30.77 kJ mol−1) and molecules kinetic diameters (0.58–0.60 nm) are almost equal [39, 40]. It is an additional proof that the kinetic stability of the inclusion compounds does not depend on the ease of the evaporation (T vap, ΔH vap) of the guest molecules [34, 35].
One can take into account that the centrosymmetrical cyclohexane molecule can easily transform from the chair conformation to the boat conformation. It will change the steric hindrance (as well as the activation barrier) for the guest molecules removal through the interconnecting channels (7.5 × 7.5 Å2) and windows (≈4 Å) of the pores.
Therefore, the entropy contribution is as favorable factor, as the energetic one in the kinetic stability of the supramolecular compounds [35].
References
Fromm KM. Coordination polymer networks with s-block metal ions. Coord Chem Rev. 2008;252:856–85.
Kirillov AM. Hexamethylenetetramine: an old new building block for design of coordination polymers. Coord Chem Rev. 2011;255:1603–22.
Mani–Biswas M, Tahir Cagin T. Insights from theoretical calculations on structure, dynamics, phase behavior and hydrogen sorption in nanoporous metal organic frameworks. Comput Theor Chem. 2012;987:42–56.
Lyszczek R, Iwan M. Investigation of desolvation process in lanthanide dinicotinates. J Therm Anal Calorim. 2011;103:633–9.
Jiang C-H, Song LF, Jiao CL, Zhang J, Sun LX, Xu F, Du Y, Cao Z. Exceptional thermal stability and thermodynamic properties of lithium based metal–organic framework. J Therm Anal Calorim. 2011;103:373–80.
Zhou YZ, Sun LX, Cao Z, Zhang J, Xu F, Song LF, Zhao ZM, Zou YJ. Heat capacities and thermodynamic properties M(HBTC)(4,4`-bipy)·3DMF (M = Ni and Co). J Therm Anal Calorim. 2012;110:949–54.
Dybtsev DN, Yutkin MP, Peresypkina EV, Virovets AV, Serre C, Ferey G, Fedin VP. Isoreticular homochiral porous metal–organic structures with tunable pore sizes. Inorg Chem. 2007;46:6843–5.
Dybtsev DN, Yutkin MP, Samsonenko DG, Fedin VP, Nuzhdin AL, Bezrukov AA, Bryliakov RP, Talsi EP, Belosludov RV, Mizuseki H, Kawazoe Y, Subbotin OS, Belosludov VR. Modular homochiral porous coordination polymers: rational design, enantioselective guest exchange sorption and ab initio calculations of host–guest interactions. Chem Eur J. 2010;10:348–56.
Yin Z, Zeng MH. Recent advance in porous coordination polymers from the view-point of crystalline-state transformation. Sci China Chem. 2011;54:1371–94.
Dyadin YuA, Soldatov DV, Logvinenko VA, Lipkovsky J. Contact stabilization of host complex molecules during clathrate formation: the pyridine–zinc nitrate and the pyridine–cadmium nitrate systems. J Coord Chem. 1996;37:63–75.
Logvinenko V, Dybtsev D, Fedin V, Drebushchak V, Yutkin M. The stability of inclusion compounds under heating. Part 2. The stability of inclusion compounds of layered zinc camphorate, linked by linear N-donor ligands. J Therm Anal Calorim. 2010;100:183–9.
Logvinenko VA, Yutkin MP, Zavakhina MS, Fedin VP. Porous metal–organic frameworks (MOFs) as matrices for inclusion compounds. Kinetic stability under heating. J Therm Anal Calorim. 2012;109:555–60.
Dybtsev DN, Chun H, Kim K. Rigid and flexible: a highly porous metal–organic framework with unusual guest-dependent dynamic behavior. Angew Chem Int Ed. 2004;43:5033–6.
Netzsch Thermokinetics. Netzsch Thermokinetics. http://www.netzsch-thermal-analysis.com/us/products-%20%20solutions/advanced-software/thermokinetics.html.
Moukhina E. Determination of kinetic mechanisms for reactions measured with thermoanalytical instruments. J Therm Anal Calorim. 2012;109:1203–14.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29:1702–6.
Friedman HL. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. J Polym Sci. 1963;6:183–95.
Ozawa T. A new method of analyzing thermogravimetric data. Bull Chem Soc Japan. 1965;38:1881–6.
Ozawa T. Estimation of activation energy by isoconversion methods. Thermochim Acta. 1992;203:159–65.
Flynn JH, Wall LA. General treatment of the thermogravimetry of polymers. J Res Nat Bur Stand. 1966;70:478–523.
Opfermann J, Kaisersberger E. An advantageous variant of the Ozawa-Flynn-Wall analysis. Thermochim Acta. 1992;203:167–75.
Opfermann JR, Kaisersberger E, Flammersheim HJ. Model-free analysis of thermo-analytical data-advantages and limitations. Thermochim Acta. 2002;391:119–27.
Vyazovkin S. Model-free kinetics: staying free of multiplying entities without necessity. J Therm Anal Calorim. 2006;83:45–51.
Simon P. Single-step kinetics approximation employing nonarrhenius temperature functions. J Therm Anal Calorim. 2005;79:703–8.
Simon P. The single-step approximation: attributes, strong and weak sides. J Therm Anal Calorim. 2007;88:709–15.
Borchard HJ, Daniels F. The application of differential thermal analysis to the study of reaction kinetics. J Amer Chem Soc. 1957;79:41–6.
Vyazovkin S, Burnham AK, Criado JM, Luis A, Perez-Maqueda LA, Popescu C, Sbirrazzuoli N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim Acta. 2011;520:1–19.
Vyazovkin S, Chrissafis K, Di Lorenzo M-R, Koga N, Pijolat M, Roduit B, Sbirrazzuoli N, Suñol J-J. ICTAC kinetics committee recommendations for collecting experimental thermal analysis data for kinetic computations. Thermochim Acta. 2014;590:1–23.
Simon P, Thomas P, Dubaj T, Cibulkova Z, Peller A, Veverka M. The mathematical incorrectness of the integral isoconversional methods in case of variable activation energy and the consequences. J Therm Anal Calorim. 2014;115:853–9.
Sestak J. Is the original kissinger equation obsolete today: not obsolete the entire non-isothermal kinetics? J Therm Anal Calorim. 2014;117:3–7.
Logvinenko V, Fedorov V, Mironov Yu, Drebushchak V. Kinetic and thermodynamic stability of cluster compounds under heating. J Therm Anal. 2007;88:687–92.
Logvinenko V. Stability and reactivity of coordination and inclusion compounds in the reversible processes of thermal dissociation. Thermochim Acta. 1999;340:293–9.
Logvinenko V. Solid state coordination chemistry. The quantitative thermoanalytical study of thermal dissociation reactions. J Therm Anal Calorim. 2000;60:9–15.
Logvinenko V. Stability of supramolecular compounds under heating. Thermodynamic and kinetic aspects. J Therm Anal Calorim. 2010;101:577–83.
Logvinenko V, Drebushchak V, Pinakov D, Chekhova G. Thermodynamic and kinetic stability of inclusion compounds under heating. J Therm Anal. 2007;90:23–30.
Pinakov DV, Logvinenko VA, Chekhova GN, Shubin YuV. The relationship between properties of fluorinated graphite intercalates and matrix composition. Part VI. J Therm Anal Calorim. 2014;115:503–9.
Logvinenko VA, Belyaev AV, Vorob’eva SN. Dehydration process of rhodium sulfate crystalline hydrate. J Therm Anal Calorim. 2013;114:1177–81.
Ukraintseva EA, Sapchenko SA, Ivanov IM, Samsonenko DG, Dybtsev DN, Manakov AY, Fedin VP. Benzene sorption by a microporous coordination polymer based on a zinc carboxilate. Rus J Inorg Chem. 2012;57:717–21.
Hauptmann S, Graefe J, Remane H. Lehrbuch der organischen Chemie. Leipzig: VEB Deutscher Verlag fur Grundstoffindustrie; 1976.
Poling BE, Prausnitz JM, O’Connell JP. The Properties of Gases and Liquids. New York: McGraw-Hill; 2001.
Acknowledgements
This work was partially supported by the Russian Foundation for Basic Research (Grant 14–03–00291).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Logvinenko, V.A., Dybtsev, D.N., Bolotov, V.A. et al. Thermal decomposition of inclusion compounds on the base of the metal–organic framework [Zn2(bdc)2(dabco)]. J Therm Anal Calorim 121, 491–497 (2015). https://doi.org/10.1007/s10973-015-4430-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-4430-6