
Abstract
The synthesis of titanium and lithium phosphates was studied using the sol-gel method. Particular attention was paid to phosphate precursors, which were mixtures of phosphate mono- and diesters prepared by solvolysis of P4O10 in isopropanol. The reaction of these precursors with titanium and lithium alkoxides yielded homogeneous gels and after drying and thermal cleavage of the esters at 300 °C, amorphous inorganic products. For the composition corresponding to the stoichiometric formula of a stable compound such as LiTi2(PO4)3, the phase crystallized as early as 550 °C by nucleation from the amorphous mixture. Ionic conductivity measured at room temperature was of the order of 10−5 S·cm−1 which increased after heat treatment at higher temperatures. If the composition did not correspond to a stable thermodynamic phase, phase separation occurred, and ionic conductivity decreased between 500 °C and 700 °C.
Graphical Abstract

Highlights
-
Lithium solid electrolyte LiTi2(PO4)3 was synthesized by sol-gel reaction.
-
Control of the reaction was performed by thermal cleavage of phosphate esters.
-
Same method was used to screen new lithium-titanium-phosphate compositions.
-
Lithium-ion conductivity was measured.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In the current state of the art, lithium batteries offer the best alternative for ensuring the autonomy of electric vehicles. However, they suffer from a number of limitations, mainly linked to their low energy-to-weight ratio and, above all, to the risks and limitations associated with the use of a liquid electrolyte (flammability, risk of dendrites and short-circuits if lithium metal is used) [1]. Many research efforts are therefore focused on the manufacture of new all-solid batteries based on the use of solid electrolytes. These batteries would deliver higher energy densities and be able to operate in wider temperature ranges [2]. However, the use of solid electrolytes raises numerous issues. Firstly, they must meet the same selection criteria as for liquid electrolytes, such as good chemical stability, a wide electrochemical stability window, a lithium transport number of 100%, and a high ionic conductivity of at least 10−4 S·cm−1 coupled to a low electronic conductivity [1]. Additionally, they must also meet requirements related to shaping and mechanical properties. Ideally, the solid electrolyte should allow easy shaping through pressing or sintering, promote a good interface between solid electrolyte grains and electrode materials, and accommodate variations in electrolyte/electrode volume during cycling. Many solutions have been proposed with oxides, sulfides, polymers, or hybrid materials [2].
Among all these materials, NASICON-type solid electrolytes substituted with lithium have the general formula LiM2(PO4)3 with M = Ti or Zr. This material is composed of phosphate (PO43−) tetrahedra linked to the transition metal (MO6) octahedra [3]. This structure features channels between these polyhedra, allowing lithium ions to circulate. The stoichiometric lithium-based material, LiTi2(PO4)3, has an interesting ionic conductivity of the order of 10−5 S·cm−1 at room temperature. However, with appropriate doping such as the substitution of M4+ by M3+, ionic conductivity can be increased up to 7 × 10−4 S·cm−1 at 25 °C [4].
Using classical solid-state reactions, it remains difficult to densify the material by sintering and conduction is limited by grain boundaries. The sol-gel method can provide an advantage in supplying reactive powders for sintering NASICON ceramics. It was first used by reacting metal alkoxides (Al, Ti, Zr…) with phosphoric acid or ammonium or alkali phosphates [5, 6]. More recently, Despandhe et al. have used sol-gel processes to obtain reactive lithium NASICON powders of composition Li1.3Al0.3Ti1.7(PO4)3 which they densified at 1000 °C to obtain high conductivities of 9 × 10−4 S·cm−1 at 25 °C [7]. They obtained a precipitate from titanium alkoxides, lithium acetate, and ammonium phosphate.
However, a precipitate is not the most suitable precursor for solid electrolyte shaping. All-solid-state batteries must be in contact with current collectors made of thin metal foils of aluminum at the cathode, and of copper at the anode. The solid electrolytes must be densified at the lowest possible temperature to avoid oxidation or melting of the metal collector. To reduce sintering temperatures, it is necessary to inhibit macroscopic precipitation. The choice of the phosphorus precursor is crucial. Phosphoric acid is a too complexing ligand and reacts too rapidly with the alkoxides of tetravalent metals (Ti, Zr) following Eqs. (1) and (2), given in the case of Ti metal.
The formation of a precipitate results from a polycondensation of titanium alkoxide with phosphoric acid, and it occurs even without hydrolysis [8]. To avoid precipitation, it seems more effective to block the phosphate reactivity by partially replacing POH functions with less reactive POR ester functions [8]. Indeed phosphate tri-esters (TEP), which do not possess any condensable POH functions neither hydrolyze nor condense. But they evaporate around 300 °C. So, in some processes described in the literature, an excess of phosphate tri-esters is required to maintain a correct stoichiometry during heating [9].
It is therefore preferred to modulate the reactivity of the phosphorus precursor by adjusting the POH/POR ratio to control the number of P–O–Ti bonds. This means using mono- and diesters of phosphoric acid, PO(OH)3-x(OR)x with x = 1 or 2, as phosphorus precursors. By controlling the POH/POR ratio, the condensation of phosphates can be limited to the formation of small condensed species. This has been demonstrated by Lugmair et al. who described the reaction of titanium isopropoxide with a diester of phosphoric acid, di-t-butylphosphate. They formed small oligomeric species (Fig. 1) [10].

Reaction of titanium isopropoxide and di-t-butylphosphate. After Lugmair and Tilley [10]
We aim at obtaining polymeric materials, that can be shaped into continuous materials without grain boundaries. In the subsequent steps of the reaction, it will still be possible to use a soft thermal cleavage of the phosphate esters at low temperatures (around 200 °C) (Fig. 2) [11]. This thermal cleavage is the result of an alcohol dehydration process. The ease of this reaction depends on the substitution level of the carbon bearing the alcohol function. A phosphate ester of a tertiary alcohol cleaves at room temperature, while that of a primary alcohol cleaves more violently at higher temperatures [8, 12].

Top: Thermal cleavage of an isopropylphosphate monoester by alcoholic dehydration, leading to the formation of phosphoric acid and propene; Bottom: Stability of phosphate esters as a function of alkoxide type, i.e. primary, secondary, or tertiary
Using phosphate ester precursors reacted with lithium and titanium alkoxides, it is then possible to obtain colloidal solutions or gels that can be deposited as thin layers and heat-treated at low temperatures to yield purely inorganic compositions. Schmutz et al. have developed such a sol-gel synthesis in an alcohol medium involving phosphate esters. Thanks to the controlled formation of P–O–Ti bonds, no precipitation was observed, and colloidal solutions were obtained that could be used for film deposition. This method was applied to the synthesis of KTiOPO4 ferroelectric material [12] as well as LiTi2(PO4)3 ion-conducting thin films onto silicon wafers [13]. A similar method was used by Takada et al. who synthesized the LiTi2(PO4)3 phase from 600 °C in thin films from lithium ethoxide, titanium isopropoxide, and phosphate esters [14]. By simply modifying the precursors, aluminum-doped Li1,3Al0,3Ti1,7(PO4)3 (LATP) phases were also produced [15]. A similar sol-gel method was used to coat powders of cathode materials (LiNi0.8Mn0.1Co0.1O2) to improve their cycling properties in lithium batteries [16].
In this work, we first revisited the use of phosphoric acid mono- and diester mixtures for the synthesis of a lithium ion-conducting material. Equimolecular mixtures of mono- and diesters were simply prepared by reacting phosphoric anhydride in an alcohol. We used isopropanol, a secondary alcohol that offers a good compromise between stability against cleavage during sol-gel condensation, and gentle thermal decomposition at around 250 °C. We then examined the mechanisms leading to the precursors decomposition to form the desired phase. We extended the method to new compositions with the aim of obtaining new crystalline phases. We then looked at the ionic conductivities of these new materials and discussed them in relation to phase separation issues.
2 Experimental
2.1 Synthesis of the mixture of phosphoric acid ester
In a typical reaction, 250 ml of isopropanol (solvent and reagent, Aldrich) were introduced into a 500 ml flask placed in an ice bath. Then 8.93 g of P4O10 (0.125 Moles of P, Aldrich) were slowly added over a period of 3 min to avoid thermal runaway. These quantities correspond to a phosphorus concentration of 0.5 mol/L. The mixture was heated under reflux for 24 h to accelerate the reaction. After the reaction, isopropanol was evaporated in a rotary evaporator under a vacuum at 50 °C. The resulting oil was dissolved again with isopropanol to form 250 mL of a solution (0.5 M in phosphorus).
2.2 Synthesis of lithium isopropoxide
Typically, 0.694 g of lithium metal (Sigma Aldrich) were added to 50 mL of isopropanol (Sigma Aldrich) in a flask. The mixture was refluxed for 24 h in a hood to remove the hydrogen formed by the reaction (Li + HOC3H7 → LiOC3H7 + ½ H2). The result was 40 g of a white solid. Assuming that no lithium was lost, the mass composition of the product was therefore 1.7% Li, but 13% molar Li, i.e. LiOR.x ROH (x = 6.55 in this case). The lithium concentration per gram was 2.5 mmol/g. In the following, we named this product solvated lithium isopropoxide and used it as is. Since it is sensitive to humidity and air, the product was stored under argon.
2.3 Synthesis of material Li/Ti/P with composition 1/2/3
First, 40 ml of a 0.5 M titanium isopropoxide solution in isopropanol (i.e. 0.02 mol Ti, Aldrich commercial) was introduced into a 200 ml flask. 4 g (0.01 mol Li) of solvated lithium isopropoxide previously synthesized were then added, and left to stir for one hour. Next, 60 ml of an equimolar mixture of 0.5 mol/L (P) phosphate esters in isopropanol (i.e. 0.03 mol P) were added. The resulting solution was stirred for 1 h before being dried in a vacuum rotary evaporator at 50 °C for 6 h, then in an oven at 100 °C for 2 h. Further heat treatments under air at various temperatures (300 °C, 500 °C, 700 °C) were also conducted.
The same procedure was used for the synthesis of other materials with adapted amounts of lithium alkoxide and phosphate esters.
2.4 Characterizations
Liquid NMR experiments were acquired on a Bruker Avance Neo 500 MHz spectrometer equipped with a BBFO Smartprobe BB&19 F/1H. All 31P and 31P{1H} NMR spectra were recorded in standard conditions at T = 25 °C in D2O, using triethylphosphate (TEP) as internal reference (δ = 0 ppm).
Solid-state 31P magic angle spinning (MAS) NMR spectra were recorded with a Bruker 500WB Avance II spectrometer (11.74T) equipped with a MAS probe of the same manufacturer. Samples were prepared in powder form and placed into 4 mm diameter zirconia rotors. Sample rotation speeds at the magic angle were maintained at 10 kHz. The 31P spectra were obtained at 202.4 MHz with a one-pulse sequence using a pulse length (π/2) of 1.32 μs, a recycle delay of 100 s, and 100 transients. For MAS NMR experiments, TEP was used as an external reference.
X-ray diffraction powder patterns were obtained with a Bruker Endeavor D8 (CoKα radiation) between 5 and 80°. Diffractograms were further simulated with the Fullprof software by Rodriguez-Carvajal [17] using Le Bail’s method. Simultaneous thermogravimetry- differential scanning calorimetry (TGA-DSC) analysis was performed on a Perkin Elmer STA 6000.
2.5 Conductivity measurements
As the materials obtained were powdery, they needed to be pressed to limit grain boundary resistance prior to the impedance measurements. For that purpose, 100 mg of product were first placed in a 1 cm diameter cell and pressed at 500 MPa. Pellets of approximately 1 mm-thick were obtained. The pellets were then placed in a Swagelok® cell between two stainless steel plungers electrically insulated from the cell body by a thin layer of Mylar polymer. The plungers’ faces were coated with conductive carbon ink to improve the contact with the sample. The assembly was tight and insulated thanks to PTFE seals tightened along the plungers by bolts (refer to Fig. 12 for details). Conductivity was determined while applying pressure on the two pistons, which act as current collectors. Indeed, pressure applied to a solid electrolyte decreases the contribution of grain boundaries by several orders of magnitude [18]. Impedance were measured between 0.1 Hz and 500 kHz using a Biologic VSP2 apparatus.
3 Results
3.1 Synthesis of the phosphate esters
The reaction schemes at the top of Fig. 3a show the species formed during the reaction between phosphoric anhydride P4O10 and excess isopropyl alcohol. The alcohol reacts with the P-O-P functions, leaving a hydroxyl function (POH) on one side and an alkoxy function (POR) on the other according to Eqs. 3 and 4:

a Representation of the intermediate steps in the reaction of P4O10 with isopropanol; b One-pulse liquid 31P NMR spectrum after 24 h reflux. In blue (top) 31P{1H} NMR and in black (bottom) 31P NMR spectra. The asterisk (*) marks the TEP reference resonance
Theoretically, the reaction results in the formation of an equimolecular mixture of monoesters and diesters. This was confirmed by the 31P NMR spectrum of the product obtained after 24 h of reflux as shown in Fig. 3b. Two major signals are observed at δ = −0.6 ppm and δ = −2 ppm. On the coupled phosphorus-proton spectrum, the peak at −0.6 ppm appears as a doublet, corresponding to the isopropylphosphate mono- ester (HO)2OPOCH(CH3)2. The 1H / 31P coupling constant is then determined at 3J = 7.6 Hz. The second peak at δ = −2 ppm is a triplet with a coupling constant 3J = 7.4 Hz, corresponding to the isopropylphosphate diester (HO)OP[-OCH(CH3)2]2. We also observed a minor singlet at δ = +1.49 ppm attributed to a trace of phosphoric acid. Integration of the peaks leads to the following molar ratio: H3PO4/monoester/diester = 2/55/43. In the following, we assumed that the mixture of phosphate esters corresponds to 1.5 POH per phosphorus atom (2 POH for a monoester and 1 for a diester). This was confirmed by acido-basic titration with sodium hydroxide in water.
3.2 Reaction of the phosphate esters with metal alkoxides
Lithium isopropoxide is not soluble in isopropanol, but it solubilizes in the presence of titanium isopropoxide. The mixture was made in ratios of Li/Ti = 0.5 to 2 from a 0.5 M titanium isopropoxide solution to which subsequent amounts of the solvated lithium isopropoxide powder (2.5 mmolLi/g) was added. Indeed, according to Kuhlman et al. [19], titanium and lithium isopropoxides form a mixed alkoxide of composition [LiTi(OiPr)5]2 soluble in isopropanol. This enabled us to obtain a homogeneous precursor solution of titanium and lithium, which we subsequently used as follows.
Phosphate esters were reacted in solution with lithium isopropoxide in LiOR/POH ratios of 0.5, 1, and 1.5. A 0.5 M solution of the phosphate monoester/diester mixture in isopropanol was mixed with the appropriate quantity of solvated lithium isopropoxide powder. Dissolution was first observed, followed by re-precipitation. 31P NMR analysis of the supernatants obtained during each of these lithiations (Fig. 4a) shows that when increasing the LiOR/POH ratio, the proportion of mono- ester was reduced relative to the one of diester. For the ratio LiOR/POH = 1, the NMR signal corresponding to the monoester completely disappeared from the supernatant. As conclusion, the monoester precipitated as a lithium salt, while the diester remained in solution. In Fig. 4a, we also show the 31P NMR spectrum coupled to 1H for LiOR/POH = 1.5 to prove that the signal at −0.3 ppm corresponds to the diesters. The characteristic triplet corresponds to the coupling of 31P with the adjacent protons of the two isopropyl groups in (HO)OP(CH(CH3)2)2. The deprotonation upon the addition of the basic lithium isopropoxide shifts its signal towards positive chemical shifts.

a 31P{1H} NMR spectra (except green one: 31P NMR spectrum) of the supernatants obtained after the reaction of lithium isopropoxide with the mixture of phosphoric acid esters; b Liquid 31P{1H} NMR spectra of a mixture of phosphate esters and titanium isopropoxide in a 1/1 P/Ti ratio
Mixing phosphate esters with titanium isopropoxide did not give rise to precipitation. However, the NMR 31P spectrum of the mixture with the ratio Ti/P = 1 showed broad peaks characteristic of oligomeric species or solid nanoparticles rapidly rotating in the solvent (Fig. 4b). The numerous peaks observed could not be precisely assigned but probably correspond to phosphates with different connectivities to titanium. Tentatively, we attribute the peak from +5 ppm to 0 ppm to phosphate esters linked to 1 titanium atom (for monoester and diester), the peaks from 0 to −5 ppm to monoesters and diesters linked to 2 titanium atoms, and the series of peaks from −10 ppm to −20 ppm to monoesters linked with 3 titanium atoms with different conformations.
As conclusion, lithium precipitated with phosphate mono- esters but the precipitation was inhibited by the presence of titanium alkoxide because of the formation of mixed lithium-titanium alkoxide and/or titanium phosphates.
3.3 Synthesis of the model ternary compound Li1Ti2(PO4)3
We first adopted this approach to synthesize a well-known ionic conductor from the literature, LiTi2(PO4)3 (LTP 1/2/3), as already discussed in the experimental part. Figure 5 shows the thermogravimetric and differential scanning calorimetry analysis of our LTP 1/2/3 material from 30 to 900 °C at 2 °C/min. The analysis was carried out under nitrogen. An initial progressive mass loss of 10% is observed between 30 and 250 °C, associated with a weakly endothermic phenomenon. This is followed by a sudden mass loss of 35% between 250 °C and 270 °C, associated with an endothermic peak at 250 °C. Finally, a small exothermic peak is observed at 550 °C, with no mass loss. X-ray diffraction analysis of the powder obtained after TGA reveals a crystalline LiTi2(PO4)3 phase. We can therefore attribute this exothermic peak at 550 °C to the crystallization of this phase from an amorphous mixture.

TGA-DSC under nitrogen flow of material obtained by mixing lithium isopropoxide, titanium isopropoxide and the mixture of phosphate mono- and diesters in the ratio Li/Ti/P = 1/2/3. Blue curve (top) represents mass (%) while black curve (bottom) shows the heat flow (mW)
The rapid mass loss at 250 °C associated with an endothermic phenomenon does not, therefore, correspond to the departure of a high-boiling molecule such as one of the phosphate esters, because the P/Ti stoichiometry would be lost in the final crystalline phase. Nor was this a calcination of organics, since the loss is endothermic and the TGA was carried out under nitrogen. This endothermic peak at 250 °C corresponds to the cleavage of phosphate esters.
If we assume cleavage by alcoholic dehydration during heat treatment, we can propose the following mechanism:
We will write R’H for propene CH2 = CH–CH3 and R for the isopropyl group CH(CH3)2.
The POH functions formed by the cleavage can react subsequently or in the same step with MOR alkoxides and release alcohols or MOH groups as well as water. For better accuracy, the ratios of phosphate mono- and diesters determined by NMR are introduced into the equation with respective percentages of 55 and 45%. This gives the following average phosphate ester composition: OP(OH)1.65(OR)1.35. The result of the thermal decomposition would be as follows.
This gives a theoretical mass loss of 52%, which is much higher than the experimental loss at 250 °C of 35% or even the total loss of 45% observed in TGA from 25 °C.
We therefore put forward a first hypothesis: during storage and handling, the residual alkoxy groups on the Li and Ti metals were hydrolyzed, giving OH rather than OR functions and leading to the formal composition LiTi2(PO4)3(R)4.05(OH)4.05.
In this case, the TGA would balance the reaction
This would give a total theoretical mass loss of 39%, this time below the total TGA loss of 45%. Thus, the white powder analyzed is the result of condensation of titanium with phosphate esters followed by a partial hydrolysis of the alkoxy groups carried by Ti and Li.
The TGA could therefore be interpreted as follows: The first progressive mass loss of 10% would be the condensation of MOH groups present and the rapid loss at 250 °C the thermal cleavage of phosphate ester functions followed by condensation with residual MOR and MOH (Fig. 6).

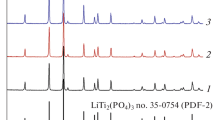
Diffractograms of materials with composition LTP 1/2/3 heated at 100, 300, 500, and 700 °C for 16 h
Solids obtained after various heat treatments were analyzed by XRD (Fig. 6). After heating at 500 °C for 16 h, the diffractogram presents diffraction peaks attributed to the LiTi2(PO4)3 crystalline phase, in addition to the broadband characteristic of an amorphous material. As a reminder, in TGA/DSC, an exothermic peak at 550 °C was attributed to material crystallization. It is therefore consistent to see a crystallized phase appearing as early as 500 °C after 16 h of heating. Heat treatment at 700 °C resulted in a diffractogram with fine, well-defined peaks corresponding to the LiTi2(PO4)3 phase. A few low-intensity peaks belong to an impurity that we have not identified. In literature, this LiTi2(PO4)3 structure is obtained by solid reaction of lithium carbonate, titanium oxide, and phosphates and requires a heating treatment at 1000 °C [20].
The diffraction pattern of the compound treated at 700 °C was simulated by Le Bail’s method, using the space group R-3c. The fit given as unit cell parameters a = 8.519 Å and c = 20.879 Å. This is in good agreement with the JCPDF 066-0871 file where unit cell parameters are given as a = 8.512 Å and c = 20.846 Å.
For comparison, a reference lithium NASICON LiTi2(PO4)3 phase was synthesized by the all-solid route [20] at 1100 °C and analyzed by X-ray diffraction. Figure 7 shows the 31P solid-state MAS NMR spectra of the various LTP 1/2/3 materials prepared at 300, 500, and 700 °C and compared with this reference NASICON structure.

One-pulse solid-state 31P MAS NMR spectra of LTP 1/2/3 materials annealed at 300, 500, and 700 °C and LTP 123 material synthesized at 1100 °C by the solid-state route. (reference of chemical shifts is TEP)
A very broad, homogeneous (unstructured) peak centered at −15 ppm is observed in the spectrum after heat treatment at 300 °C. This is characteristic of an amorphous material with a wide distribution of environments.
From 500 °C, two fine peaks appear at −27 ppm and −29 ppm, as well as a broad peak from −10 to −35 ppm. At 700 °C, the fine peak at −27 ppm becomes the major contribution, with two small peaks between −2 and −8 ppm, which are probably related to impurities. The material synthesized by a solid route at 1100 °C shows a single peak at −27 ppm, which is characteristic of phosphorus in the NASICON crystal structure environment, i.e. a phosphate connected to 4 titanium atoms [21].
We conclude that after thermal cleavage, we obtain a homogeneous amorphous phase. The coordination of phosphates abruptly changes around 500 °C by nucleation of an ordered structure with well-defined connectivity, identical to that of the NASICON structure.
4 Exploration of new compositions
In the remainder of this work, we investigated the possibility of obtaining metastable phases of various compositions by this low-temperature synthesis. The working hypothesis was that amorphous mixtures may prefer to crystallize a metastable phase with a local ordering rather than to separate into more stable phases as this process would require a long-range diffusion process which is unfavorable at low temperatures. This was called diffusion-limited- crystallization by Lange et al. [22, 23]. Indeed, a low-temperature process may enable us to obtain homogeneous inorganic mixtures as early as 300 °C. We, therefore, sought to obtain orthophosphates (formed from PO43− ions) compositions with the general formula LixTiy(PO4)z. This implies that the compositions verify the electroneutrality relationship with Li+, Ti4+, and PO43− ions:
Several compositions were selected based on this affine function, featuring higher [Li]/[P] ratios than the NASICON composition. They are referenced in Fig. 8 where compositions already reported in the literature are marked with a blue triangle. They are Ti3(PO4)4 [24], LiTi2(PO4)3, Li5Ti(PO4)3 [25] and Li3PO4. Compositions marked with a red cross, on the other hand, correspond to unknown compositions that we tested.

Representation of tested orthophosphate compositions with the general formula LixTiy(PO4)z with respect to electroneutrality
The material in this family with the highest Li/P ratio is lithium phosphate (Li3PO4), well known in the literature. This material is a lithium salt with very low ionic conduction (10−7 S·cm−1) [26]. However, the most conductive materials in the literature (such as Li10GeP2S12) are derived from this structure by substituting oxides for sulfides and phosphorus for tin and germanium [27]. Ionic conductivity may be related to the volume density of lithium in the material. We therefore looked for compositions richer in lithium than lithiated NASICON but less than basic lithium phosphate, in the hope that titanium would stabilize the structure (against reaction with CO2 for example).
The synthesis method for these compositions was the same as that described above for LTP 1/2/3. In the case of Li3PO4 (LTP 3/0/1), a precipitate was obtained, but the supernatant-precipitate combination was dried and then reground to homogenize. In all other cases, homogeneous solutions were obtained yielding transparent powders after removal of the isopropanol solvent with a rotary evaporator.
Every sample was first characterized by TGA (heating rate 5 °C, under nitrogen (Fig. 9). All of them gave similar weight losses from 170 °C to 650 °C. However, when the material composition was more acidic (high [Ti]/[P] ratio), the mass loss occurred at a lower temperature: it occurred at 310 °C for LTP 3/0/1 and decreased down to 250 °C for LTP 1/2/3. Moreover, this loss was also more abrupt in that case. Cleavage was nevertheless gentle since an endothermic peak was systematically observed on the DTA curves (not shown here).

Thermogravimetric analysis of LTP compositions 1/2/3, 2/1/2, 12/3/8, 5/1/3, and 3/0/1. For better comparison, the mass of the materials at 650 °C was taken as a reference. The ordinate should be understood as a difference of mass losses, taking as a reference the masses of the materials obtained at 650 °C
All systems presented a similar behavior, and we will only focus on the composition LTP 2/1/2 as an example. Figure 10 shows the diffractograms obtained after heat treatments carried out at temperatures between 100 and 700 °C for 24 h on the LTP 2/1/2 composition. All solids dried at 100 °C were amorphous. However, after heat treatment at 300 °C, a crystalline phase of γ-Li3PO4 appeared in addition to the amorphous compound. The solid had thus undergone phase dissociation, with lithium reacting with the surrounding phosphates to form a lithium phosphate.

X-ray diffractograms of materials with composition LTP 2/1/2 after heating at 100, 300, 500, and 700 °C respectively
At 500 °C, the Li3PO4 crystalline phase could still be observed, but the LiTi2(PO4)3 crystalline phase appeared as well. Indeed, if we add the two compositions LiTi2(PO4)3 and Li3PO4 in equal parts, we obtain LTP 4/2/4, i.e. the composition of LTP 2/1/2. This gives the phase separation estimated by reaction (9). At 700 °C, the two crystalline structures are still present, but the diffraction peaks are thinner than at 500 °C.
The LTP 2/1/2 material was also studied with solid 31P MAS NMR. Figure 11 shows the spectra of heat-treated LTP 2/1/2 materials at 300, 500, and 700 °C. As early as 300 °C, a complex spectrum is observed. It is structured into several unresolved peaks whose maxima would be at +10, +2, −5, −10, and −15 ppm. In contrast with LTP 1/2/3, this indicates a local phase separation. The fine peak at +10 ppm can be attributed to γ-Li3PO4. Indeed, the same signal was obtained on commercial γ-Li3PO4 (Sigma). NMR confirms the X-ray diffraction conclusions that at 300 °C at least two phases are obtained, one of which is crystallized Li3PO4 and the other an amorphous Li-Ti-P mixture.

One-pulse solid-state 31P MAS NMR spectra of materials with composition LTP 2/1/2 after heating at 300, 500, and 700 °C respectively
At 500 °C, the Li3PO4 signal was still observed at −10 ppm. The broad massif barycenter shifts towards negative displacements and widens to −30 ppm, revealing two new environments at −23 ppm and −29 ppm.
From 700 °C onwards, one sees an intense peak at −9 ppm and another at −27 ppm characteristic of the NASICON LTP 1/2/3 structure. It is therefore possible that Li3PO4 reacted with the amorphous phases to give rise, this time by solid reaction, to a new phase, which, however, could not be identified on the XRD diagram.
MAS NMR revealed the formation of crystallized Li3PO4 at low temperatures by phase separation of an amorphous phosphate phase. At higher temperatures (700 °C), the lithium phosphate formed a new phase, which we were unfortunately unable to further study. Longer syntheses at 700 °C or higher temperatures would have been necessary.
In summary, solution synthesis produced homogeneous amorphous mixtures, but it was not possible to crystallize a metastable complex phase locally from this composition, as a separation with the stable Li3PO4 phase occurred as soon as the phosphate ester functions were cleaved at around 250 °C.
4.1 Conductivity measurements
Materials with various compositions were studied in terms of impedance spectroscopy and their resistance was measured. Figure 12 shows the impedance diagrams (Nyquist plots) measured at 25 °C under a pressure of 100 MPa for the composition LTP 2/1/2 heat-treated at 300, 500, and 700 °C. The impedance diagrams show two circles that are classically associated with grain boundary contribution at low frequency and the intrinsic conductivity of the grains at high frequency. However, we observed that the size of both circles depends on the applied pressure, showing an intricated mixture of both contributions. Still, we considered the resistance of the material (including grain boundaries) and the intercept of the high-frequency circle with the x-axis.

Nyquist plots (impedance diagrams) for LTP 2/1/2 obtained at 300, 500, and 700 °C. Conductivities were measured at 20 °C under a pressure of 100 MPa
The conductivity results are shown in Fig. 13. All the phases obtained at 500 °C have a conductivity of the order of 10−6 S·cm−1. The LTP 1/2/3 composition was the only one that showed an increase of its conductivity between the sample treated at 500 °C and 700 °C, probably because no phase separation occurred, and the material was better crystallized. For the other compounds, conductivity peaked after treatment at 500 °C. After heat treatment at 700 °C, their conductivity decreased, and this is probably associated with phase decomposition. Phase dissociation at higher temperatures probably led to the formation of insulating or poorly conducting phases (Li3PO4 or impurities) that act as a barrier to conduction between the conducting LiTi2(PO4)3 grains.

Conductivity of the LTP with different compositions after heat -treatment at 300 °C, 500 °C, and 700 °C. Measurements made at 25 °C under a 100 MPa pressure applied to the current collectors
5 Conclusion
In this work, we have confirmed the feasibility of solid electrolyte synthesis from phosphate esters and metal alkoxides. The mixtures obtained were homogeneous and yielded homogeneous materials at room temperature. In this way, we obtained materials that were easy to condense - via cold-pressing processes. Their endothermic decomposition in a non-oxidizing atmosphere enabled us to limit runaway during synthesis and control their formation and sintering.
A known phase of lithium NASICON was synthesized in this way at a low temperature (700 °C), giving the same ionic conductivities as those reported in the literature for materials obtained by the solid route at a higher temperature (1000 °C). A perspective would be to study the effect of doping with aluminum and to study the shaping of these gels into thin films or coatings.
The method enabled us to obtain gels of varying compositions of lithium/titanium/phosphate. However, from the lowest temperatures (300 °C), a phase separation was observed between the Li3PO4 compound and an amorphous material. At higher temperatures, only the most stable and well-known phases crystallized.
Powders prepared at 500 °C all had a conductivity of 10−6 S·cm−1, but only the composition corresponding to the LiTi2(PO4)3 crystalline phase showed an improvement of its conductivity after treatment at higher temperatures.
References
Goodenough JB, Kim Y (2010) Challenges for rechargeable Li batteries. Chem Mater 22(3):587–603. https://doi.org/10.1021/cm901452z
Kim JG, Son B, Mukherjee S, Schuppert N, Bates A, Kwon O, Choi MJ, Chung HY, Park S (2015) A review of lithium and non-lithium based solid state batteries. J Power Sources 282:299–322. https://doi.org/10.1016/j.jpowsour.2015.02.054
Goodenough JB, Hong HY-P, Kafalas JA (1976) Fast Na+-ion transport in skeleton structures. Mater Res Bull 11(2):203–220. https://doi.org/10.1016/0025-5408(76)90077-5
Fu J (1997) Fast Li+ ion conduction in Li2O-Al2O3-TiO2-SiO2-P2O2 glass-ceramics. J Am Ceram Soc 80(7):1901–1903. https://doi.org/10.1111/j.1151-2916.1997.tb03070.x
Perthuis H, Colomban P (1986) Sol-gel routes leading to nasicon ceramics. Ceram Int 12(1):39–52. https://doi.org/10.1016/S0272-8842(86)80008-6
Bucharsky EC, Schell KG, Hintennach A, Hoffmann MJ (2015) Preparation and characterization of sol–gel derived high lithium ion conductive NZP-type ceramics Li1+x AlxTi2−x(PO4)3. Solid State Ion 274:77–82. https://doi.org/10.1016/j.ssi.2015.03.009
Deshpande A, Bansod S (2024) Effect of sintering temperature on sol-gel synthesized nasicon-type Li1.3Al0.3Ti1.7(PO4)3 ceramic solid electrolyte. J Mater Sci Mater Electron. https://doi.org/10.1007/s10854-023-11766-z
Livage J, Barboux P, Vandenborre MT, Schmutz C, Taulelle F (1992) Sol-gel synthesis of phosphates. J Non-Cryst Solids 147–148:18–23. https://doi.org/10.1016/S0022-3093(05)80586-1
Vasiliu I, Gartner M, Anastasescu M, Todan L, Predoana L, Elisa M, Negrila C, Ungureanu F, Logofatu C, Moldovan A, Birjega R, Zaharescu M (2007) Structural and Optical Properties of the SiO2–P2O5 Films Obtained by Sol–Gel Method. Thin Solid Films 515(16):6601–6605. https://doi.org/10.1016/j.tsf.2006.11.106
Lugmair CG, Tilley TD (1998) Di- Tert- butyl phosphate complexes of titanium. Inorg Chem 37(8):1821–1826. https://doi.org/10.1021/ic971347e
Paciorek KJL, Kratzer RH, Kaufman J, Nakahara JH, Christos T, Hartstein AM (1978) Thermal oxidative degradation studies of phosphate esters. Am Ind Hyg Assoc J 39(8):633–639. https://doi.org/10.1080/0002889778507827
Schmutz C, Basset E, Barboux P, Maquet J (1993) Study of titanium phosphate gels and their application to the synthesis of KTiOPO4 films. J Mater Chem 3(4):393. https://doi.org/10.1039/jm9930300393
Schmutz C, Basset E, Barboux P (1993) Couches minces de phosphates de titane par voie sol-gel. J Phys III 3(4):757–766. https://doi.org/10.1051/jp3:1993161
Takada K, Fujimoto K, Inada T, Kajiyama A, Kouguchi M, Kondo S, Watanabe M (2002) Sol- Gel Preparation of Lithium Ion Conductive Thin Film. Appl Surf Sci 89:300–306. https://doi.org/10.1016/S0169-4332(01)01007-8
Kunshina GB, Gromov OG, Lokshin EP, Kalinnikov VT (2014) Sol-gel synthesis of Li1.3Al0.3Ti1.7(PO4)3 solid electrolyte. Russ J Inorg Chem 59(5):424–430. https://doi.org/10.1134/S0036023614050118
Qu X, Yu Z, Ruan D, Dou A, Su M, Zhou Y, Liu Y, Chu D (2020) Enhanced electrochemical performance of Ni-rich cathode materials with Li 1.3 Al 0.3 Ti 1.7 (PO 4) 3 coating. ACS Sustain Chem Eng 8(15):5819–5830. https://doi.org/10.1021/acssuschemeng.9b05539
Rodriguez-carvajal J (1993) Recent advances in magnetic structure determination neutron powder diffraction. Phys B 192:55–69
La Monaca A, Girard G, Savoie S, Demers H, Bertoni G, Krachkovskiy S, Marras S, Mugnaioli E, Gemmi M, Benetti D, Vijh A, Rosei F, Paolella A (2021) Effect of pressure on the properties of a NASICON Li1.3Al0.3Ti1.7(PO4)3 nanofiber solid electrolyte. J Mater Chem A 9(23):13688–13696. https://doi.org/10.1039/D1TA01143J
Kuhlman R, Vaartstra BA, Streib WE, Huffman JC, Caulton KG (1993) Primary steps in the hydrolyses of two heterometallic alkoxides. Characterization of lithium-titanium and barium-zirconium isopropoxides [LiTiO(O-Iso-Pr)3]4 and BaZr4(OH)(O-Iso-Pr)17. Inorg Chem 32(7):1272–1278. https://doi.org/10.1021/ic00059a040
Cretin M, Fabry P, Abello L (1996) ChemInform Abstract: Li1+xAlxTi2-x(PO4)3 for Li+ potentiometric sensors. ChemInform. https://doi.org/10.1002/chin.199606010.
Arbi K, Kuhn A, Sanz J, García-Alvarado F (2010) Characterization of lithium insertion into NASICON-type Li1 + x Ti2 − x Al x (PO4) 3 and its electrochemical behavior. J Electrochem Soc 157(6):A654. https://doi.org/10.1149/1.3368764
Lange FF, Balmer ML, Levi CG (1994) Diffusion limited crystallization and phase partitioning in ZrO2-metal oxide binary systems: code: D3. J Sol Gel Sci Technol 2(1–3):317–321. https://doi.org/10.1007/BF00486263
Barboux P, Griesmar P, Ribot F, Rolles L (1995) Homogeneity-related problems in solution derived powders. J Solid State Chem 117(2):343–350. https://doi.org/10.1006/jssc.1995.1283
Ponnala B, Balla P, Hussain SK, Ginjupalli SR, Koppadi K, Nekkala N, Perupogu V, Lassi U, Seelam PK (2022) Selective hydrogenolysis of biodiesel waste bioglycerol over titanium phosphate (TiP) catalysts: the effect of Pt & WO3 loadings. Waste Biomass- Valoriz 13(11):4389–4402. https://doi.org/10.1007/s12649-022-01909-4
Rao KJ, Sobha KC, Kumar S (2001) Infrared and Raman spectroscopic studies of glasses with NASICON-type chemistry. J Chem Sci 113(5–6):497–514. https://doi.org/10.1007/BF02708786
Ayu NIP, Kartini E, Prayogi LD, Faisal M, Supardi (2016) Crystal structure analysis of Li3PO4 powder prepared by wet chemical reaction and solid-state reaction by using X-ray diffraction (XRD). Ionics 22(7):1051–1057. https://doi.org/10.1007/s11581-016-1643-z
Kanno R, Murayama M (2001) Lithium ionic conductor Thio-LISICON: the Li[Sub 2]S-GeS[Sub 2]-P[Sub 2]S[Sub 5] system. J Electrochem Soc 148(7):A742. https://doi.org/10.1149/1.1379028
Acknowledgements
NMR equipment at ESPCI Paris PSL is funded in part by the Paris Region. We thank Agence Nationale de la Recherche et de la Technologie for the financial support of A. Guillot during thesis contract CIFRE n°2020/1330.
Author information
Authors and Affiliations
Contributions
AG performed the synthesis characterization during his PhD work, helped by AS during her master’s internship. MNR performed the liquid-state NMR characterizations at Chimie Paristech and J-BD the solid-state NMR experiments at ESPCI. CM and XR supervised the work on the industrial site while DG and PB supervised the PhD and academic work. AG and PB prepared the figures. PB, AG, and DG wrote the main manuscript text. All authors reviewed the document.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guillot, A., Giaume, D., Suvorova, A. et al. Synthesis of lithium conducting titanium phosphates by the sol-gel process. J Sol-Gel Sci Technol 111, 336–346 (2024). https://doi.org/10.1007/s10971-024-06461-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-024-06461-2