
Abstract
Zn(OAc)2(H2DEA) was synthesized by the reaction of zinc acetate dihydrate (Zn(OAc)2•2H2O) with diethanolamine (H2DEA), and was characterized using single-crystal X-ray structural analysis, nuclear magnetic resonance spectroscopy, Fourier-transform infrared (FT-IR) spectroscopy, and elemental analysis. Zn(OAc)2(H2DEA) had a trigonal bipyramidal geometry comprised of one zinc atom, two acetate groups, and one H2DEA as a neutral tridentate ligand to form two five-membered rings. The states of Zn(OAc)2(H2DEA) heated at various temperatures were determined by FT-IR spectroscopy. At 270 °C, the H2DEA ligand dissociated and was removed. The absorption bands assigned to Zn–O stretching vibration of Zn4O core such as the zinc-oxo cluster appeared. When heated at 500 °C, the absorption bands of μ4-oxozincate and the acetate group disappeared completely and hexagonal wurtzite structural ZnO was formed at 550 °C. A possible thermal decomposition pathway from Zn(OAc)2(H2DEA) to ZnO was proposed. The ZnO film was highly transparent and formed by the deposition of ZnO nanoparticles with size ~40 nm.

Zn(OAc)2(H2DEA) was synthesized and characterized. The states of Zn(OAc)2(H2DEA) heated at various temperatures were determined by FT-IR spectroscopy, and the formation mechanism of ZnO was estimated.
Highlights
-
Zinc–diethanolamine complex was synthesized by the reaction of zinc acetate with diethanolamine.
-
Zinc–diethanolamine complex was isolated and characterized.
-
The formation mechanism of ZnO was estimated by FT-IR spectra.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Zinc oxide, ZnO, has attracted attention for its application in sensors, transparent conductors, etc. [1,2,3,4,5,6,7]. ZnO films are prepared by sputter deposition, chemical vapor deposition, sol–gel process, etc. [3, 8, 9]. The sol–gel process is a good technique because it is an easy process, of low cost, and easily coats large surface areas. Therefore, many researchers prepare ZnO films using the sol–gel process. ZnO precursor is prepared by the sol–gel reaction of zinc salts (e.g., zinc acetate, zinc nitrate, and zinc alkoxides) with alkanolamines such as monoethanolamine (HMEA), diethanolamine (H2DEA), and triethanolamine (H3TEA). ZnO thin films are deposited by spin-coating or dip-coating on a substrate followed by heat treatment [2, 10,11,12,13,14,15,16,17].
The formation mechanism of ZnO suggests that ZnO colloids or gels are formed by the polymerization of zinc hydroxide and/or zinc-oxo acetate clusters from ZnO precursors [12, 17]. The structures of ZnO precursors were proposed by Znaidi and Nehmann. Znaidi et al. [12, 13] proposed that Zn–monoethanolamine and Zn acetate complexes were present in equilibrium when mixing Zn(OAc)2 and HMEA. Nehmann et al. [18] proposed that the Zn–diethanolamine complex Zn(DEA) was formed as a ZnO precursor by ligand exchange reaction between Zn(OAc)2 and H2DEA. However, these ZnO precursors could not be isolated and characterized and the authenticity of ZnO precursors remains uncertain. Recently, Conterosito et al. [19] reported the synthesis of the Zn–triethanolamine complex Zn(OAc)2(H3TEA) by mixing Zn(OAc)2 with triethanolamine (H3TEA). The complex was characterized by single-crystal X-ray structural analysis and transferred to cluster compounds (Zn4(OAc)4(HTEA)2) by adding water. One of the zinc–alkanolamine complexes was thus revealed.
In this work, Zn–diethanolamine complex, which is useful and is one of the zinc–alkanolamine complexes, was synthesized by the reaction of Zn(OAc)2•2H2O with diethanolamine (H2DEA) in EtOH (Scheme 1). The Zn–diethanolamine complex Zn(OAc)2(H2DEA) was isolated and characterized by single-crystal X-ray structure determination, nuclear magnetic resonance (NMR) spectra, Fourier-transform infrared (FT-IR) spectrum, and thermal analysis. Moreover, the formation mechanism of ZnO through the complex was studied via FT-IR spectra.

Synthesis of Zn(OAc)2(H2DEA)
2 Experimental section
2.1 Reagents
The solvents Zn(OAc)2•2H2O and H2DEA were purchased from Wako Pure Chemical Industries Ltd., Tokyo, Japan, and used as received. Zn4(μ4-O)(OAc)6 was prepared according to the literature [20], and the method and characterization were described in supporting information. Silicon wafer (4” polishing wafer) was purchased from GlobalWafers Co., Ltd. The substrate was cut to 2.5 cm × 2.5 cm and cleaned with a neutral detergent, EtOH, and ethyl acetate.
2.2 Measurements
All NMR spectra were recorded on a JEOL JNM-ECP500 spectrometer (JEOL, Akishima, Japan; 1H at 500.16 MHz, 13C at 125.77 MHz) at ~23 °C. The 1H NMR and 13C{1H} NMR spectra were recorded using tetramethylsilane as internal standard. FT-IR spectra were recorded on a JASCO FT/IR-6100 spectrometer (JASCO, Hachioji, Japan) using the KBr method or after coating on silicon wafer. Elemental analysis was performed using a Perkin Elmer 2400II Elemental Analyzer. Melting points were recorded using a Bibbly Stuart Scientific SMP3 instrument; the reported melting points were uncorrected. Thermogravimetric-differential thermal analysis (TG-DTA) was performed using a NETZCH JAPAN TG-DTA 2000SE (Netzsch Japan, Yokohama, Japan). Samples were heated to 800 °C at the rate of 10 °C/min under airflow. X-ray diffraction (XRD) spectra were recorded using an IC Vario (PANAlytical, Tokyo, Japan) with monochromatic Cu-Kα radiation as X-ray source. Atomic force microscope (AFM) observations were obtained using an SPM-9700 (Shimadzu, Kyoto, Japan). Transmittance spectra were recorded using a JASCO V-670 spectrophotometer equipped with an integrating-sphere photometer ISN-470 type (JASCO, Hachioji, Japan) in the wavelength range of 300–800 nm by coating films on glass substrates.
Crystal data were collected using a Bruker AXS SMART APEX CCD X-ray diffractometer equipped with monochromatic Mo-Kα radiation (0.7107 Å). Empirical absorption corrections using equivalent reflections and Lorentzian polarization corrections were performed using the SADABS program [21]. All data were collected with SMART and Bruker SAINTPLUS (Version 6.45) software packages. The structures were solved using the SHELXA-97 program [22] and refined against F2 using SHELEXL-97 [23].
The deposition number was CCDC-1567248 for Zn(OAc)2(H2DEA). Free copies of the data can be obtained via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK; Fax:+44 1223 336033; e-mail: deposit@ccdc.cam.ac.uk).
2.3 Synthesis of Zn(OAc)2(H2DEA)
Zn(OAc)2•2H2O (0.66 g, 3.0 mmol) and H2DEA (0.31 g, 3.0 mmol) were dissolved in 5 mL of EtOH, and the mixture was refluxed overnight. The solution was concentrated to about half the volume, and kept standing at −18 °C for 1 h. A white powder was obtained from the solution by reprecipitation with hexane. The obtained powder was purified by recrystallization with tetrahydrofuran (THF), and Zn(OAc)2(H2DEA) was obtained as colorless crystals (0.50 g, 69%).
M.p. (dec.) = 130.4–132.5 °C. Elemental analysis: calcd. for C8H17NO6Zn C 33.29, H 5.94, and N 4.85; found C 33.66, H 5.86, and N 4.90. 1H NMR (500 MHz, CD3OD): δ = 1.96 (s, 6 H, CH3), 2.95 (t, J = 5.0 Hz, 4H, CH2–N), 3.74 (t, J = 5.0 Hz, 4H, O–CH2), and 4.91 (s, 3H, NH and OH) ppm. 13C{1H} NMR (126 MHz, CD3OD): δ = 23.01 (CH3), 50.05 (CH2–N), 59.51 (O–CH2), and 181.01 (COO) ppm. FT-IR (KBr, cm−1): 3412, 2969, 2889, 1577, 1409, 1341, 1099, 1057, 1023, 933, 899, 678, and 619.
2.4 Preparation of ZnO thin film
ZnO thin film was prepared by spin-coating (500 rpm for 10 s followed by 1000 rpm for 40 s) using 10 wt% Zn(OAc)2(H2DEA)/EtOH solution (200 μL) dropped on a substrate and drying at 100 °C for 10 min. This process was repeated twice. The coating film was heated at various temperatures, at the rate of 10 °C/min, followed by keeping at a chosen temperature for 30 min.
3 Results and discussion
3.1 Synthesis of Zn(OAc)2(H2DEA)
Zn(OAc)2(H2DEA) was synthesized by the reaction of Zn(OAc)2•2H2O with H2DEA in EtOH, and purified by reprecipitation with hexane followed by recrystallization from hot THF. After 1 d, single crystals of Zn(OAc)2(H2DEA) appeared. Zn(OAc)2(H2DEA) was easily soluble in alcohol and hot THF, and insoluble in hexane, toluene, and chloroform. The structure of Zn(OAc)2(H2DEA) was stable even in the presence of a small amount of water, unlike that of Zn(OAc)2(H3TEA).
3.2 Characterization of Zn(OAc)2(H2DEA)
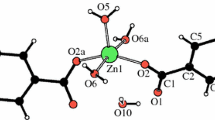
The structure of Zn(OAc)2(H2DEA) was measured by single X-ray structural analysis, as shown in Fig. 1. Zn(OAc)2(H2DEA) had a trigonal bipyramidal geometry comprised of one zinc atom, two acetate groups, and one H2DEA as a neutral tridentate ligand to form two five-membered rings (Fig. 1a). The distances of these bonds were Zn1–O1 (2.176(3) Å), Zn1–O2 (2.202(3) Å), Zn1–O3 (1.990(3) Å), Zn1–O4 (1.988(3) Å), and Zn1–N1 (2.059(4) Å); therefore, these bonding distances increased in the order Zn–OAc < Zn–N of H2DEA < Zn–O of H2DEA. The structural geometry, bonding distances, and angles of Zn(OAc)2(H2DEA) were similar to that of Zn(OAc)2(H3TEA) [19]. Interestingly, Zn(OAc)2(H2DEA) showed the formation of hydrogen bonding between one central complex and three complexes, and the hydrogen atoms in H2DEA interacted with acetate groups of other complexes (Fig. 1b). These hydrogen bond distances were in the order O–H…O = C (1.792 Å) < O–H…O–Zn (1.918 Å) < N–H…O = C (2.294 Å).

ORTEP drawings of a the molecular structure of Zn(OAc)2(H2DEA) and b interaction with thermal ellipsoids at 50% probability level. Crystal solvents are omitted for clarity. Color code: gray, H; white, C; red, O; blue, N; light blue, Zn
1H and 13C NMR spectra of Zn(OAc)2(H2DEA) were obtained using CD3OD. For both the NMR spectra, the signals assigned to H2DEA ligand shifted slightly downfield compared to that of H2DEA. In the FT-IR spectrum, the absorption bands were assigned to vO–H and vN–H at 3200–3500 cm−1, vC–H at 2969 and 2889 cm−1, vC = O at 1577 cm−1, vCOO at 1409 cm−1, δC–H at 1340 cm−1, ρC–H at 1023 cm−1, δCOO at 670 cm−1, πCOO at 617 cm−1 [24, 25], vC–N at 1099 cm−1 [26], vC–O at 1060 and 991 cm−1 [26, 27], and τO–H and τN–H at 899 cm−1 [26].
3.3 Thermal analysis and FT-IR spectra of Zn(OAc)2(H2DEA)
The thermal analysis of Zn(OAc)2(H2DEA) was operated by TG-DTA in airflow, as shown in Fig. 2. The temperature at 134 °C appeared as an endothermic peak and weight loss was observed. A small endothermic peak appeared at 263 °C, and the weight loss value was 20.6%. The temperature was similar to the boiling point of H2DEA. Exothermic peaks due to oxidation reaction appeared at 310, 387, 414, and 467 °C with weight losses of 19.0, 14.1, 8.8, and 7.2%, respectively. Finally, the organic compounds were completely burnt, and ZnO was obtained as char ceramic (char yield 27.2%, calculated yield 28.2%). The obtained ZnO was derived from the wurtzite structure by the XRD spectrum (see Supporting information).

TG-DTA traces of Zn(OAc)2(H2DEA) under airflow
The states of Zn(OAc)2(H2DEA) at various temperatures were measured by FT-IR spectra, as shown in Fig. 3. The measured samples were prepared using 100 μL of 10 wt% Zn(OAc)2(H2DEA)-ethanolic solution on silicon wafer by spin-coating at various temperatures for 30 min in air. At 110 °C, the IR spectrum was similar to that of pure Zn(OAc)2(H2DEA). When heated at 140 °C, the band assigned to vCOO at 1407 cm−1 was slightly broader than that at 110 °C. From 1H NMR spectrum, the signals of methylene derived from H2DEA appeared at two regions comprising different chemical environments, and the endothermic peak for TG-DTA appeared at 134 °C. Therefore, the temperature was identified with the decomposition temperature of Zn(OAc)2(H2DEA). On additional heating to 270 °C, the bands assigned to vN–H, vC–H, and vC–O (at 3414 and 3246, 2935 and 2866, and 1060 cm−1, respectively) almost disappeared. The broad band at 1566 cm−1 overlapped with the bands due to δO–H at 1623 cm−1 and vC = O at 1562 cm−1; therefore, the H2DEA ligand was completely dissociated and evaporated at 270 °C. Moreover, a new band at 516 cm−1 appeared. The new band was assigned to Zn–O stretching vibrations derived from cluster-forming Zn4O cores such as Zn4O(OAc)6 [28, 29]. However, the IR spectrum was different from that of pure Zn4O(OAc)6 (see Supporting information). Probably, the coating film contained a segment of μ4-oxozincate formation. When heated to 420 °C, the bands assigned to vC=O and vC–O disappeared, and vZn–O was unclear due to cleavage of the Zn4O structure. Moreover, the band due to vO–H was assigned to the absorption of water and/or hydroxyl groups of Zn compounds. Finally, the bands of organic, hydroxyl groups, and Zn4O core completely disappeared at 500 °C.

FT-IR spectra of Zn(OAc)2(H2DEA) on heat treatment at a 110 °C, b 140 °C, c 270 °C, d 420 °C, and e 500 °C for 30 min
Based on report [12], we propose the formation of ZnO through Zn(OAc)2(H2DEA) as shown in Scheme 2. When heated at 140 °C, a part of the coordinated hydroxyl group on H2DEA was dissociated and/or recoordinated to other zinc atoms as in polymerization (intermediate 1). At 270 °C, most of the H2DEA was removed and Zn(OAc)2 changed to ZnO colloidal formation from a few μ4-oxozinc compounds such as Zn4O(OAc)6, Zn10O4(OAc)12, and Zn5(OH)8(OAc)2•2H2O because the band at 516 cm−1 was assigned to Zn–O stretching vibrations of the Zn4O core and these compounds were prepared by the hydrolysis–condensation of Zn(OAc)2 in the presence of a basic compound [30,31,32]. After heating at 420 °C, the acetato groups were burnt and the hydroxyl group was formed; at 500 °C, ZnO was formed by dehydration–condensation between hydroxyl groups. From the result, it was clear that H2DEA acted as a ligand and also as a basic compound to form μ4-oxozinc compounds.

A possible thermal decomposition pathway from Zn(OAc)2(H2DEA) to ZnO
3.4 Characterization of ZnO thin film via heat treatment of Zn(OAc)2(H2DEA)
The ZnO thin film was prepared by heat treatment at 550 °C of Zn(OAc)2(H2DEA) coated on a glass substrate. The ZnO thin film was characterized by XRD and AFM, as shown in Fig. 4. The XRD pattern of ZnO thin film appeared at the diffraction peaks of (100), (002), (101), (102), and (110) as derived from the hexagonal wurtzite structure [33, 34]. A broad peak between 20° and 40° was assigned to the amorphous glass substrate. Densely packed ZnO particles were observed with a size of approximately 40 nm using AFM. Finally, the transmittance was high (>90%) in the visible region.

a XRD spectrum and b AFM images of ZnO thin film
4 Conclusion
Zn(OAc)2(H2DEA) was synthesized by the reaction of Zn(OAc)2•2H2O with H2DEA in EtOH. The structure of the intermediate was revealed. It was characterized by single-crystal X-ray structural analysis, NMR and FT-IR spectra, and elemental analysis. The single-crystal X-ray structure comprised a zinc atom, two acetate groups, and one H2DEA as a neutral ligand. From the TG-DTA and FT-IR study, Zn(OAc)2(H2DEA) decomposed at 140 °C. At 270 °C, most of the H2DEA was removed and μ4-oxozinc compounds were formed. After heating at 420 °C, the acetato groups burned to form the hydroxyl group. At 500 °C, ZnO was formed. The ZnO film formed from Zn(OAc)2(H2DEA) showed high transparency (>90%) and a hexagonal wurtzite structure. Densely packed ZnO particles were observed with a size ~40 nm.
References
Fortunato E, Barquinha P, Martins R (2012) Oxide semiconductor thin-film transistors: a review of recent advances. Adv Mater 24:2945–2986
Tari O, Aronne A, Addonizio ML, Daliento S, Fanelli E, Pernice P (2012) Sol–gel synthesis of ZnO transparent and conductive films: a critical approach. Sol Energy Mater Sol Cells 105:179–186
Sirelkhatim A, Mahmud S, Seeni A, Haida N, Kaus M, Ann LC, Bakhori SKM, Hasan H, Mohamad D (2015) Review on zinc oxide nanoparticles: antibacterial activity and toxicity mechanism. Nano-Micro Lett 7:219–242
Hussain M, Tahir MN, Mansoor MA, Arifin Z, Mazhar M (2013) Heptanuclear zinc cluster for growth of zincite and manganese-doped zincite thin films for sensor applications. Mon Chem 144:285–294
Liu Y, Li Y, Zeng H (2013) ZnO-based transparent conductive thin films: doping, performance, and processing. J Nanomater 2013:196521
Rodnyi PA, Khodyuk IV (2011) Optical and luminescence properties of zinc oxide. Opt Spectrosc 111:776–785
Adl AH, Kar P, Farsinezhad S, Sharma H, Shankar K (2015) Effect of sol stabilizer on the structure and electronic properties of solution-processed ZnO thin films. RSC Adv 5:87007–87018
Stadler A (2012) Transparent conducting oxides—an up-to-date overview. Materials 5:661–683
Kolodziejczk-Radzimska A, Jesionowski T (2014) Zinc oxide—from synthesis to application: a review. Materials 7:2833–2881
Khatibani AB, Abbasi M (2018) Effect of Fe and Co doping on ethanol sensing property of powder-based ZnO nanostructures prepared by the sol–gel method. J Sol–Gel Sci Technol 86:255–265
Li H, Wang J, Liu H, Yang C, Xu H, Li X, Cui H (2004) Sol–gel preparation of transparent zinc oxide films with highly preferential crystal orientation. Vacuum 77:57–62
Znaidi L, GJAAS Illia, Benyahia S, Sanchez C, Kanaev AV (2003) Oriented ZnO thin films synthesis by sol–gel process for laser application. Thin Solid Films 428:257–262
Znaidi L, GJAAS Illia, Guennic RL, Sanchez C, Kanaev A (2003) Elaboration of ZnO thin films with preferential orientation by a soft chemistry route. J Sol–Gel Sci Technol 26:817–821
Chen WJ, Liu WL, Hsieh SH, Hsu YG (2012) Synthesis of ZnO:Al transparent conductive thin films using the sol–gel method. Proc Eng 36:54–61
Salam S, Islam M, Akram A (2013) Sol–gel synthesis of intrinsic and aluminum-doped zinc oxide thin films as transparent conducting oxides for thin film solar cells. Thin Solid Films 529:242–247
Ohya Y, Saiki H, Takahashi Y (1994) Preparation of transparent, electrically conducting ZnO film from zinc acetate and alkoxide. J Mater Sci 29:4099–4103
Znaidi L (2010) Sol–gel-deposited ZnO thin films: a review. Mat Sci Eng B-Solid 174:18–30
Nehmann JB, Ehrmann N, Reineke-Koch R, Bahnemann DW (2014) Aluminum-doped zinc oxide sol–gel thin films: influence of the sol’s water content on the resistivity. Thin Solid Films 556:168–173
Conterosito E, Croce G, Palin L, Boccaleri E, van Beekab W, Milanesio M (2012) Crystal structure and solid-state transformations of Zn–triethanolamine–acetate complexes to ZnO. Cryst Eng Comm 14:4472–4477
Gordon RM, Silver HB (1982) Preparation and properties of tetrazinc μ4-oxohexa-μ-carboxylates (basic zinc carboxylates). Can J Chem 61:1218–1221
Sheldrick GM (1996) SADABS, Program for Simens area detector absorption correction, University of Göttingen, Germany
Sheldrick GM (1997) SHELXS-97, Program for crystal structure solution, University of Göttingen, Germany
Sheldrick GM (1997) SHELXL-97, Program for crystal structure refinement, University of Göttingen, Germany
Johnson MK, Powell DB, Cannon RD (1981) Vibrational spectra of carboxylato complexes—I. Infrared and Raman spectra of beryllium(II) acetate and formate and of zinc(II) acetate and zinc(II) acetate dihydrate. Spectrochim Acta A Mol Spectrosc 37:899–904
Zhang Y, Zhu F, Zhang J, Xia L (2008) Converting layered zinc acetate nanobelts to one-dimensional structured ZnO nanoparticle aggregates and their photocatalytic activity. Nanoscale Res Lett 3:201–204
Masoud MS, El-Enein SAA, Abed IM, Ali AE (2002) Synthesis and characterization of aminoalcohol complexes. J Coord Chem 55:153–178
Brannon DG, Morrison RH, Hall JL, Humphrey GL, Zimmerman DN (1971) Spectra and bonding for copper(II)-aminoalcohol complexes—I: the I.R. spectra of complexes of mono-, di- and triethanolamine. J Inorg Nucl Chem 33:981–990
Berkesi O, Dreveni I, Andor JA, Goggin PL (1991) Formation and mid-FT-IR investigation of short (C2–C5) straight chain tetrazinc μ4-oxohexa-μ-carboxylates. Inorg Chim Acta 181:285–289
Spanhel L (2006) Colloidal ZnO nanostructures and functional coatings: a survey. J Sol–Gel Sci Technol 39:7–24
Meulenkamp EA (1998) Synthesis and growth of ZnO nanoparticles. J Phys Chem B 102:5566–5572
Tokumoto MS, Pulcinelli SH, Santilli CV, Briois V (2003) Catalysis and temperature dependence on the formation of ZnO nanoparticles and of zinc acetate derivatives prepared by the sol–gel route. J Phys Chem B 107:568–574
Schmidt T, Müller G, Spanhel L (1998) Activation of 1.54 μm Er3+ fluorescence in concentrated II–VI semiconductor cluster environments. Chem Mater 10:65–71
Viart N, Richard-Plouet M, Muller D, Pourroy G (2003) Synthesis and characterization of Co/ZnO nanocomposites: toward new perspectives offered by metal/piezoelectric composite materials. Thin Solid Films 437:1–9
O’Brien S, Nolan MG, Çopuroglu M, Hamilton JA, Povey I, Pereira L, Martins R, Fortunato E, Pemble M (2010) Zinc oxide thin films: characterization and potential applications. Thin Solid Films 518:4515–4519
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “New Polymeric Materials Based on Element-Blocks” (No. 2401) (JSPS KAKENHI Grant Number JP24102008). This work was also supported by JSPS KAKENHI Grant Number JP16K17951.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Hayami, R., Endo, N., Abe, T. et al. Zinc–diethanolamine complex: synthesis, characterization, and formation mechanism of zinc oxide via thermal decomposition. J Sol-Gel Sci Technol 87, 743–748 (2018). https://doi.org/10.1007/s10971-018-4768-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-018-4768-x