
Abstract
This work presents a detailed investigation about the influence of a complementary set of imidazolium ionic liquids (IL: [C4MIm][BF4]–[C10MIm][BF4]–[C7O3MIm][BF4]–[C4MIm][NTf2]–[C10MIm][NTf2]–[C7O3MIm][NTf2]) in the preparation of hydrolytic sol–gel silica wet gels and xerogels, providing insights into their hierarchical structures, morphologies of primary particles and aggregation domains. The reported IL decreased dramatically the gelation time (up to ~500 times), evolving the systems to transparent or opalescent wet gels with distinct consistencies, or silica powders. Characterization of the obtained silica xerogels was performed by transmission and scanning electron microscopy, atomic force microscopy, X-ray diffraction, small-angle X-ray scattering and thermogravimetric analysis. In general, the IL allowed controlling the silica particle size, color, compactness, structures and morphology. The xerogels showed homogenous and ordered structures (spherical or triangular) of different sizes and with distinct microporosities, depending on the IL applied. These results highlight the key role of IL in the sol–gel silica synthesis with controlled properties, which can be further tuned for specific applications by changing small process parameters.
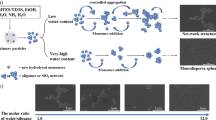
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Inorganic–organic hybrid materials produced via the sol–gel process are fascinating materials that allow the permanent incorporation of organic groups into the inorganic system, integrating the properties of both components. Moreover, the sol–gel process enables the preparation of multifunctional materials with high purity and homogeneity at relatively mild conditions, turning this technique widely applied in fields like coatings [1], optics [2], biohybrids [3], drug delivery [4] and the preparation of nanoparticles, including nanosilica [5]. Intrinsically, the nanoscale corresponds to an elevated active surface area, which increases the number of reactive centers (Si–OH), enhancing the adsorption ability as well as chemical reactivity [6]. Those characteristics make this type of silica highly desirable in a wide variety of fields, e.g., catalysis [7], biotechnology [8] and nanocomposites [9].
The most common synthetic method for silica is based on hydrolyzable semi-metal alkoxide precursors like the commonly used, low-cost, tetraethyl orthosilicate [TEOS, Si(OC2H5)4] [10]. This entire process is highly dependent on several parameters like temperature, solvent, pH [11, 12], presence of ions [11], air moisture [13], drying process and aging time [14]. The impact caused by changes in these parameters on the competing hydrolysis and condensation reactions, and as a consequence on the final products, is still not fully understood. On the other hand, this method’s flexibility allows designing the material’s bulk properties, e.g., molded gels [15], mesoporous spun fibers and hollow spheres [16], thin films [12], nanotubes [17], molecular cages [18], aerogels and xerogels [5], when establishing the parameter–property rationale.
Ionic liquids (IL) are organic salts that are in the liquid state at temperatures of up to 100 °C and belong to a class of multifunctional surfactants. Most of these ionic–covalent molecules present negligible vapor pressures, high electrical conductivity and heat capacity, non-flammability under ambient conditions [19] and promising antimicrobial [20, 21], antitumoral and antioxidative properties [22], which make them very attractive for a constantly increasing number of application fields [23]. IL form extended hydrogen bond networks in both solid and liquid phases, together with high ionic mobility and stability in the presence of air and moisture. Structural alterations in the hydrophobic and hydrophilic IL regions cause a reorganization of its ions, optimizing the intermolecular forces for each modification. Such small changes, e.g., differences in alkyl imidazolium side-chain length, ether-functionalization or anion exchange, cause variations in viscosity, ionic conductivity [24] and solubility, as well as shifts in degradation and glass transition temperatures [19, 25].
Innovative hybrid materials can be synthesized via the silica sol–gel method using IL as either solvent [26, 27], co-solvent [5] or template [28–32]. This in situ application of IL into sol–gel silica formation processes allows structure control, driven by the IL’s self-assembling property and selective IL–substrate interactions while preserving their specific properties. IL interact with the growing particles through the hydrogen bond “co-π–π stacking” mechanism [30], enabling the formation of new organic–inorganic multifunctional materials in one-pot procedures under mild reaction conditions. This creates an ordered IL solvation layer on the silica surface, which influences the surface interactions. Thus, differences in the size, geometry, polarity and Coulomb coupling forces between anions and cations contribute directly to the final silica particle size, compactness and morphology. Previously, we reported on the role of in situ applied 1-monoethylene glycol monomethyl ether-3-methylimidazolium [C3O1MIm] cation-based IL on the morphology of sol–gel silica xerogels. This modification led to highly distinct silica xerogel morphologies dependent on the IL anion, forming compact lamellar monoliths with the [MeSO3] anion, free flowing powders when using the [BF4] anion and porcelain-like aggregates while applying the [PF6] anion [33]. This class of materials can be explored in fields like electrochemistry [34], catalysis [35] and nanocomposites. Both the ex situ melt blending of IL–silica xerogel hybrids with thermoplastics [36] and the in situ formation of IL–silica–thermosetting polymer nanocomposites [37] significantly improve thermo-mechanical properties of polymer matrices. In those systems, the IL functioned as multifunctional compatibilizing agents, affecting the silica particle size, size distribution and morphology, and the final properties of these nanocomposites [38].
In this work, the in situ application of small IL contents as multifunctional components in the synthesis of silica xerogels as potential low-cost nanofillers is described. Particular effects of different IL with diverse cation–anion combinations on gelation times, thermal degradations, compactness, structures and morphologies were investigated.
2 Experimental
2.1 Materials
1-n-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide [C4MIm][NTf2], 1-n-butyl-3-methylimidazolium tetrafluoroborate [C4MIm][BF4], 1-n-decyl-3-methylimidazolium tetrafluoroborate [C10MIm][BF4] and 1-n-decyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide [C10MIm][NTf2] were purchased from IoLiTec Ionic Liquids Technologies Inc. The procedure reported previously in the literature was used for the synthesis of 1-triethylene glycol monomethyl ether-3-methylimidazolium tetrafluoroborate [C7O3MIm][BF4], and the 1H-NMR spectral data confirmed its identity and purity, considering the absence of the characteristic [MeS] anion peak [39]. 1-Triethylene glycol monomethyl ether-3-methylimidazolium bis(trifluoromethylsulfonyl)amide [C7O3MIm][NTf2] was also prepared by anion exchange using a 1:1 molar ratio of 1-triethylene glycol monomethyl ether-3-methylimidazolium methanosulfonate [C7O3MIm][MeS] and lithium bis(trifluoromethylsulfonyl)amide (LiNTf2). After stirring for 2 days in dichloromethane, the reaction mixture was filtered to remove LiMeS and dried under reduced pressure for solvent removal. The 1H-NMR spectral data of [C7O3MIm][NTf2], presented in the Supplementary Information (Fig. SI1), confirmed its chemical structure and the complete exchange of the [MeS] anion. Before application into the sol–gel systems, the IL were dried under vacuum for 5 h at 60 °C. The silica precursor tetraethoxysilane (TEOS, Sigma-Aldrich), HCl (37 %, Sigma-Aldrich) and EtOH, Absolut P.A. (VETEC Química Fina LTDA) were used without further purification. The PERMUTION® column system was used for water deionization.
2.2 In situ preparation of silica xerogel
The IL–silica xerogel hybrids were prepared through an adapted literature procedure for the synthesis of silica xerogels in the presence of IL [33, 40]. The molar ratios of n EtOH/n TEOS, n H2O/n TEOS and n IL/n TEOS were fixed at 5.6, 2.9 and 0.03, respectively. In order to evaluate the temperature influence, the reaction was conducted at room temperature (RT) and at 60 °C. The 0.01 M HClaq solution with a pH of 2.0 was prepared by dilution of 0.83 mL HCl (12 M) in 1000 mL deionized water. This pH value was proposed by Letailleur et al. [12] to promote rapid hydrolysis and cluster–cluster growth to form stable, homogeneous silica thin films. The IL (type and mass specified in Table 1), EtOH (15.0 g) and TEOS (12.2 g) were mixed until homogenous (~5 min), followed by the addition of HClaq (3.1 g). This reaction mixture was stirred during the sol formation (~20 min), left to rest in a closed reaction vessel at RT or 60 °C, allowing silica self-organization, until wet gel formation. Gelation time (t gel) was determined for each reaction, defined as the moment when the sample did not exhibit flow in an angled reaction vessel [41]. Due to the continuation of the condensation reaction after the gel point [11], the wet gels were left to age for 7 days at RT. This step was introduced to decrease the chance of intense aging during drying, which promotes agglomeration [42]. The gels were then dried under vacuum for solvent removal (5 h at 60 °C), and photographic images were taken of the resulting silica xerogels, which were grinded using ceramic mortar and once more dried (5 h at 60 °C) before further analyses (TEM, SEM, TGA and SAXS). Modified silica xerogels obtained at 60 °C were additionally calcinated for 3 h at 600 °C in a muffle furnace for X-ray diffraction (XRD) analyses.
For the reader’s clearer understanding, short-hand notations have been used for differentiating xerogel silicas from pure IL: IL-modified silicas are denoted by the straightforward IL abbreviation (e.g., C4MImBF4), while square brackets have been applied for pure IL to separate its cation and anion (e.g., [C4MIm][BF4]).
2.3 Silica xerogel nanoparticles characterization
2.3.1 Transition electron microscopy (TEM)
The specimen’s morphologies and structures were examined by TEM using a JEOL JEM-1200 Ex II operating at an accelerating voltage of 80 kV. Silica xerogels were first dispersed in deionized water under sonication for 20 min and deposited on 200-mesh Formvar/carbon-coated Cu grids.
2.3.2 Scanning electron microscopy (SEM)
The specimen surface was analyzed by SEM using the EVO® 50—Carl Zeiss operating at 5 kV. Each sample was placed on a sample holder covered with a PELCO Carbon Conductive Tab, and the excess of powder was gently removed. The samples were sputtered with platinum to increase their electric conductivity.
2.3.3 Atomic force microscopy (AFM)
AFM images of silica surfaces were obtained in the tapping mode using Nanoscope IIIa manufactured by Digital Instruments Co. Scans were performed with the NANOSENSORS™ probe made of silicon nitride (model: PPP-NCl-50) operating at 154.7 kHz frequency, 1 line/second scanning speed, 0° angle, 256 points/line resolution and set point of ~3.1 V. The WSxM 6.0 software from Nanotec Electronic S.L. was used for the image treatments. Samples for AFM were prepared as thin films on microscope glass slides via dip-coating in hydrolyzed sols and subsequent condensation in a closed vessel.
2.3.4 Thermogravimetric analyses (TGA)
TGA were performed using a TA Instruments QA-50. The IL–silica xerogel hybrids and pure IL were heated from 25 to 900 and 25 to 700 °C, respectively, at a heating rate of 10 °C/min. All experiments were conducted under nitrogen atmosphere with a gas flow rate of 60 mL/min. The sample (~10.0 mg) was placed in a platinum crucible, and an empty platinum pan was used as reference for all measurements. The temperature of highest degradation rate (T d) was determined, using the maximum temperature peak of the first TGA derivative curve (DTG). Photographic pictures of the residual ashes were taken after the analyses.
2.3.5 X-ray diffraction (XRD)
The XRD experiments were carried out on a SIEMENS D500 diffractometer equipped with a curved graphite crystal as monochromator, using Cu Kα radiation (λ = 1.5406 Å). The diffraction data were collected at RT in Bragg–Brentano θ–2θ geometry. The equipment was operated at 40 kV and 17.5 mA with scan ranges of 1°–45° and 0.2°–5°. The diffractograms were obtained with a constant step of Δ2θ = 0.05 and Δ2θ = 0.02 for the wide and small angle ranges, respectively. For analysis, samples in the powder form were evenly spread on the ground glass support. The Bragg equation nλ = 2dsinθ was applied for interplanar distance (d) calculations.
2.3.6 Small-angle X-ray scattering (SAXS)
SAXS analyses were performed using an Anton Paar SAXSess camera equipped with a 2D imaging plate detector. Cu Kα X-rays of 1.5418 Å wavelength were generated by a Philips PW3830 sealed tube source (40 kV, 50 mA) and slit-collimated. Grinded silica powder was placed between two polyimide sheets before obtaining spectra. All scattering data were corrected for the background and normalized for the primary beam intensity. In order to remove the inelastic scattering from the data, the SAXS profiles were additionally corrected for both Porod constant and desmearing effect.
3 Results and discussion
The sol–gel technique was applied to prepare a total of twelve IL–silica xerogel hybrids (Table 1), considering the complementary set of six imidazolium IL presented in Fig. 1 and at two different reaction temperatures, RT and 60 °C. These imidazolium IL have been identified as excellent electrolytes for electrochemical applications. Since the addition of even small amounts of electrolytes into the hydrolytic sol–gel process can change the evolution of particles sizes and morphologies [11], Rahman et al. [43] studied the role of ammonium salts’ anions when introduced into this silica formation process. They proposed that the silica nanoparticle sizes formed are related to the anions’ size. Karout and Pierre [5] showed that the addition of imidazolium IL increased the gelation time and, by changing the IL concentrations, it was possible to control the xerogel and aerogel silica pore sizes. Moreover, in previous publications from our group [33, 38], both IL’s ionic counterparts dramatically affected the sol–gel process. Thus, in this study, a combination of three different imidazolium cation side-chains and two different anions allowed investigating the roles of the anions and cations sizes, geometries, polarities and the anion–cation Coulombic forces, from the hybrids’ formation process to their final structural, morphological and thermal properties.

Room temperature imidazolium IL applied in this work: [C4MIm][BF4]–[C10MIm][BF4]–[C7O3MIm][BF4]–[C4MIm][NTf2]–[C10MIm][NTf2]–[C7O3MIm][NTf2]
3.1 IL–silica gelation time
The gelation process depends on adhesive interparticle interactions [44] causing the sol–gel transition kinetics to be tunable by manipulating different reaction parameter values [11] and/or by the addition of a modifier [9]. Thus, by applying IL, TEOS hydrolytic polycondensation is strongly affected, especially due to different IL’s acidity and polarity profiles [33, 40]. Based on that, the gelation time (t gel) and the visual aspects of wet gels were investigated (Table 1). Interestingly, the determined t gel shows a general trend for all samples: IL promote the gelation process in the acid (HCl)-catalyzed sol–gel system when present from the beginning. Although this goes in opposition to the Karout and Pierre results [5], their systems involved the application of IL only after the alkoxysilane pre-hydrolysis, showing the important role of IL in catalyzing and organizing the sol–gel hydrolysis step, influencing the entire process, as previously demonstrated by our group [45]. As for the temperature dependency, t gel followed an Arrhenius behavior for all samples. Increasing the temperature from RT to 60 °C (by ~40 °C), the t gel was reduced by ~11 times for the samples C4MImNTf2, C7O3MImNTf2 and C10MImNTf2; ~20 times for C10MImBF4; and ~24 times for C4MImBF4. For C7O3MImBF4, this comparison was not possible, as it did not reach the t gel at 60 °C.
When compared to the IL-free reference samples, C4MImBF4 decreased its t gel dramatically at both 60 °C (~500 times) and RT (~300 times). Also for C10MImBF4, the t gel was reduced by ~300 (60 °C) and ~250 times (RT), and C7O3MImBF4, prepared at RT (t gel not reached at 60 °C), presented a t gel shortened by ~40 times. In general, [NTf2] anion-based IL showed only a moderate effect on the t gel, showing at most half of the original t gel values.
The influence of cations with similar chain length, but different polarity (C10MIm vs. C7O3MIm), on the t gel was evaluated. The most polar IL, containing the [C7O3MIm] cation and [BF4] anion, increased the t gel of the system at RT. Differently, at 60 °C it caused slow (after ~24 h) and gradual phase separation, as a result of a phenomenon referred as “creaming,” which occurs when the formed species present lower density than the dispersing fluid and they tend to rise to the surface [46]. In the case of this system, the Brownian motion seems to surpass the interparticle interaction and gravity effects, preventing this system from reaching a gelation point and forming a very small and free flowing powder stable into the solvent. For C7O3MImNTf2, compared to its aliphatic analog (C10MImNTf2), the t gel at both temperatures decreased (by 30 % at RT and 25 % at 60 °C).
The differences in gelation rates could be related to the aggregation mechanism; e.g., [BF4] acts as basic catalyst, causing monomer–cluster aggregation and fast condensation, while [NTf2] functions as acid catalyst, causing cluster–cluster aggregation and slow condensation. The anion reactivity could also have a considerable contribution to t gel. The [BF4]-based IL are reported to react exothermally with water, which could both increase locally the solution temperature, or even undergo hydrolysis producing hydrogen fluoride (HF) [47, 48]. Both temperature and HF (a nucleophilic catalyst for the process) would result in speeding up gelation. For the [NTf2] anion, the negative charge is dispersed across the S–N–S moiety, which turns it much more stable in water, showing slower condensation in comparison with the less stable [BF4] anion [47]. Considering the above-mentioned, in order to predict the t gel it is necessary to evaluate a complex variety of IL properties, including the IL’s reactivity in the medium, as well as their polarities and cation–anion interaction strengths.
3.2 Morphology
3.2.1 Wet gels
At the beginning of the sol–gel hydrolysis, all systems, including the IL-free wet gels, presented homogeneous, colorless and transparent appearance, most likely, due to slow condensation rates [49]. The C4MImBF4 and C10MImBF4 sols became lightly opalescent after 15 min (60 °C) or 2 h (RT) of reaction, keeping this aspect until gelation. After reaching the gelation point, lightly opalescent (RT) and opalescent (60 °C) wet gels were formed (Fig. 2). Opalescence occurs when material scatters the shorter wavelengths of the visible spectrum, giving the material a bluish hue under reflected light. This may indicate the existence of ordered clusters made of silica nanoparticles, where the size of the clusters would be similar to the wavelength of the incident light, acting as scattering centers. Kozuka and Sakka [50] observed the same phenomenon for gels presenting shorter t gel and higher HCl content. In the case of C7O3MImBF4, no gel structure was formed when synthesized at 60 °C, while at RT formed a lightly opalescent gel. This behavior could be associated with the medium viscosity and condensation kinetic differences due to the temperature change. [C7O3MIm][BF4] presents the highest viscosity, hydrophilicity, hydrogen-bonding capability and polarity among the used IL, due to the synergistic existence of both the ether-functionalized cation and the anion of higher symmetry [24]. Since the temperature applied was not high enough to break H-bonding formed between silanols and both IL cation and anion, a decreased medium and IL viscosities probably caused phase separation. For C4MImNTf2, C10MImNTf2 and C7O3MImNTf2, the twice-shorter t gel did not result in changes in the visual wet gels appearance in comparison with IL-free samples. Comparing the visual appearance with t gel of the studied systems, shorter t gel results in opalescent wet gels while longer t gel in transparent ones. This indicates two anion-dependent growth mechanisms of aggregation: (1) predominating cluster-cluster growth, resulting in a transparent homogenous gel formed by smaller aggregates of bigger particles, for the [NTf2] anion, and (2) monomer–cluster aggregation, or at least its significant contribution, resulting in fast condensation and an opalescent gel formed by large aggregates of smaller particles, for the [BF4] anion. Moreover, the visual aspect is normally correlated to the network bonding, which also affects the gel hardness. IL-free wet gels presented very low densities and were “soft to touch,” in opposition to all in situ IL-modified wet gels, which were much stiffer. C4MImBF4 and C10MImBF4 showed the highest stiffness, suggesting the formation of a higher number of cross-links, which normally results in the stiffening and strengthening of the network [11].

Photographic images of in situ IL-modified wet gels, prepared under identical conditions using different IL, formed at 60 °C. From the left to the right: transparent C4MImNTf2 and C10MImNTf2, followed by opalescent C4MImBF4 and C10MImBF4
3.2.2 Xerogels
The xerogel silica prepared from the previously discussed wet gels presented striking contrasts in volumes, colors and textures, depending principally on the IL applied (Fig. 3). When identical mass fractions (1 g) were compared, drastic variations in the volume were observed. For the samples prepared at RT, the most pronounced visual differences were regarding the coloration of the different silicas, while the volumes were relatively proportional (Fig. 3a). On the other hand, the role of the IL anion on the final silica structure became evident for the silicas prepared at 60 °C, where systems based on [BF4] IL produced volumes about twice larger than IL-free or [NTf2] IL-based ones (Fig. 3b). All samples turned into white, free flowing powders after grinding.

Comparison of 1 g non-grinded silica xerogels synthesized under identical conditions at either RT (a) or 60 °C (b), varying only the IL applied. From the left to right: IL-free, C4MImBF4, C10MImBF4, C7O3MImBF4, C4MImNTf2, C10MImNTf2 and C7O3MImNTf2
The xerogels’ microscopic images provided an insight into the particles sizes, structures, surfaces and organization. Herein, a coupled imagery study was organized to evaluate the different parameters for system organization. The TEM images were obtained from solution-dispersed silica (synthesized at 60 °C), giving information about the adhesion among clusters. The AFM images were obtained from microscope glass slides dip-coated into the sols and casted under controlled environment, evaluating the influence of casting or concentrating of the reaction mixture during the curing process at RT. The SEM images were recorded directly from the grinded xerogels (obtained at 60 °C), giving an idea about the systems surface when produced under a closed environment, keeping the solvent volume constant during the whole process.
IL-free samples resulted in highly aggregated and compact particles with broad size distribution (Fig. 4a). This agglomeration can be ascribed to van der Waals forces, electrostatic interactions and hydrogen bonding between –OH groups, intensified by the presence of the catalyst [11]. Moreover, in aqueous systems, hydrodynamic and capillary effects during the drying process also promote agglomeration [9]. The condensation kinetics played an important role in defining the sizes of both xerogels’ primary particles and aggregates. The fast gelating systems (C4MImBF4 and C10MImBF4) developed small nanoparticles; i.e., the size of primary particles was only a few nm, forming low compactness aggregates (Fig. 4b, c, respectively) together with loose spherical particles on the surface (Fig. 4c). Systems with slower condensation rates (C4MImNTf2 and C7O3MImNTf2) formed larger spherical particles (~50 nm) (Figs. 4d, 5d). Surprisingly, although C10MImNTf2 also presented a slow condensation rate, much smaller particles were developed, despite the similar monolith aggregation characteristic presented for all three [NTf2]-based IL (Fig. 4e).

TEM images with inserted AFM topology scans of a IL-free, b C4MImBF4, c C10MImBF4, d C4MImNTf2 and e C10MImNTf2 xerogels. c, e additionally include SEM images of corresponding silica xerogels obtained at 60 °C

TEM micrographs with inserted AFM scans of a, b C7O3MImBF4 and c, d C7O3MImNTf2 obtained at RT and 60 °C, respectively
These broad system variations could have their origin in the hydrogen-bonding (H-bond) formation between IL anions and Si–OH groups, as well as in the strength of those interactions. The stability of this H-bond depends mainly on the size, geometry and Coulomb coupling forces between the IL’s anions and cations [33, 38] (Scheme 1). During hydrolysis, a series of dynamic interactions take place, completely changing the course of the condensation process. Together with the H-bond on the growing silica surface, IL tend to self-assemble in solution [51] forming an ordered solvation layer and decreasing the chance of silica interparticle random aggregation. As a result, the silica domains have to grow through the “unprotected paths” (Scheme 1). Based on this hypothesis, a bulkier anion should produce a more tortuous path for the silica domain to grow, producing more linear and smaller aggregates. This can be observed by TEM for xerogels modified with [NTf2]-containing IL, which tend to form “crescent-shape”-like structures (Figs. 4d, 4e, 5d), probably shaped by the geometry of its bulky anion (Scheme 1d). The morphology changes were also captured by AFM, and the images are presented as inserts in the corresponding TEM images.

Representation of IL dynamic templating for samples presenting stronger H-bond: a C10MImBF4 and b C7O3MImBF4, and weaker H-bond: c C10MImNTf2 and d C7O3MImNTf2, between Si–OH and IL’s anion
Regarding the change in the synthesis temperature, considerable differences in particles morphologies have been identified for the C7O3MImBF4 and C7O3MImNTf2 xerogels. The C7O3MImBF4 network formation at RT resulted in more homogenous and much smaller particles (Fig. 5a) than the “creamed” particles prepared at 60 °C (Fig. 5b). Interestingly, the C7O3MImNTf2 xerogel formed at RT presented “corn grain-like” structures (Fig. 5c) and spherical particles when performed at 60 °C (Fig. 5d). Primarily, the main driving force for these changes could be ascribed to the ether-functionalized IL’s capacity to form multiple H-bonding interactions [24, 45]. This would allow the formation of metastable aggregates that organize the initial system, strongly influencing the structure and morphology of the final product. The solvent regimen during the condensation step was identified as another crucial “synergistic” aspect for the system organization. Systems prepared for AFM via dip-coating, despite presenting the same basic characteristics, are in general smaller and much more defined than the ones prepared under a constant solvent concentration. This is best exemplified by the C7O3MImNTf2 hybrid prepared at RT (Fig. 5c), which presents rough but good correlation of its triangular or “corn grain-like” structures for both TEM and AFM. The particles in AFM images are half the size of those in TEM micrographs and present a much more homogeneous morphology and size distribution. This indicates that system concentration during hydrolysis enhances the IL multiple H-bond effect, making the templating more defined than with constant solvent concentration.
3.3 Structural organization
XRD analyses were performed to investigate the structural organization of the silica xerogels synthesized at 60 °C, before and after calcining (Fig. SI2). Table 2 presents the recorded diffraction angles and calculated d values, only for samples that present a non-amorphous peak. All the broad peaks at 2θ 1 = ~23° are characteristic of amorphous systems with short range ordering. In general, the calcination process slightly shifted this peak to lower 2θ values, indicating modification of the silica networks related to increased d values [52]. The IL-free, C4MImNTf2 and C10MImNTf2 (plateau) systems showed a second peak at 2θ 2 = ~6°, corresponding to the existence of long range ordering that can be ascribed to the acid-catalyzed induced microporosity [11]. This peak disappeared after calcination, indicating the loss of the silica’s microporosity. In the case of C7O3MImNTf2, this reflection was shifted to 2θ 2 = ~2° after calcination, which corresponds to an increase in the pore size. The C4MImBF4 and C10MImBF4 systems presented shifts to regions of smaller angles even before calcination, which could be explained by the change of an acid-catalyzed mechanism to an acid–base-catalyzed one, which induced the formation of larger particles and interparticle distances [11]. The reminiscence of this peak indicates a more stable network formation even after templates’ removal, or even integration of the decomposition products to the silica network.
Silica xerogels modified with ether-functionalized IL, prepared at RT and 60 °C, were also investigated by SAXS analysis. The SAXS plots in terms of I(q) versus scattering vectors (q p ) are presented in Fig. 6a, b. Derived values from the slopes in fractal (−G) and Porod (−P) linear regions (Fig. SI3), as well as the mean size of primary particles (α = 2π/q P ) and surface fractals dimensions (d s = 6 − P) [53], are gathered in Table SI1. As mentioned above, acid-catalyzed sol–gel systems have intrinsic inhomogeneities in the form of microporosity. Herein, the presence of microporosity is confirmed by the existence of a plateau in I(q) [54], especially for samples prepared at 60 °C (curves 2, 4, 6). The P values indicate the presence of primary particles with coarse surfaces (P value ≥−3). Samples prepared at 60 °C (curves 2, 4, 6) present a larger number of smaller particles (higher intensity in the range of q between 1 and 3 nm), which build fractal aggregates with lower fractal density (lower G) when compared to RT samples (curves 1, 3, 5). The same sample prepared at RT (curve 5) formed ~5 times smaller elementary particles, without microporosity plateau, and more loose aggregates of compact particles. This decrease in compactness was also observed in SEM images (Fig. 6c, d). By the simple anion exchange, it was possible to obtain a variety of SAXS fractal profiles that confirm the direct IL interaction with growing silica systems.

SAXS profiles: a including the microporosity region, b Porod plot after subtraction of Porod constant, arrow represents the position of the scatter vector q P from the fractal to the Porod region from IL-free (1 and 2), C7O3MImNTf2 (3 and 4) and C7O3MImBF4 (5 and 6) systems obtained at RT and 60 °C, respectively. SEM micrographs of the C7O3MImBF4 xerogels showing the compactness change when gelated at RT (c) or “creamed” at 60 °C (d)
3.4 Thermal stability
IL thermal resistance measurements were taken for pure IL (Fig. 7a, b) and for silica-confined IL (Fig. 7c, d), and the results are summarized in Table 1. The T d of pure IL was well defined, presenting sharp events. All IL with the [NTf2] anion displayed better thermal resistances than those with the [BF4] anion. The longer imidazolium cation alkyl chain decreased the IL thermal stability, in accordance with the literature [55]. [C4MIm][NTf2] (curve 4) had the highest thermal stability and [C7O3MIm][BF4] (curve 3) the lowest, with an expressive difference of 50 °C. This behavior is normally observed for ether-functionalized IL when compared to aliphatic analogs [24]. Surprisingly, [C7O3MIm][NTf2] (curve 6) did not follow this tendency and presented a T d in the same range as its aliphatic analog. This indicates that the NTf2 anion’s positive influence on total thermal resistance superposes that of the negatively influencing ether cation side-chain. Additionally, [C7O3MIm][NTf2] and [C10MIm][NTf2] were not completely decomposed when reaching 700 °C, indicating the formation of thermal-resistant IL’s decomposition products.

Representative TGA and DTG curves a, b of pure IL (1) [C4MIm][BF4], (2) [C10MIm][BF4], (3) [C7O3MIm][BF4], (4) [C4MIm][NTf2], (5) [C10MIm][NTf2] and (6) [C7O3MIm][NTf2] and c, d the 60 °C silica-confined IL (1′) C4MImBF4, (2′) C10MImBF4, (3′) C7O3MImBF4, (4′) C4MImNTf2, (5′) C10MImNTf2 and (6′) C7O3MImNTf2
The xerogel hybrids, despite the peak corresponding to the vaporization of strongly physisorbed water molecules (below 220 °C), presented characteristic IL degradation events (Fig. 7d). The intensity contrast between DTG curves is related to the IL mass percentage difference in xerogels, especially for samples with different anion (Table 1). Superimposed DTG/TGA curves were used for the quantitative determination of IL in the hybrids. Approximate IL contents were determined by subtracting the mass % at the end (m %B ) from mass % at the beginning of the DTG peak (m %A ) and associating it with the total mass applied in the analysis (Eq. 1). The obtained average mass values of IL are close to the ones predicted theoretically (Table 1; Eq. 2). Due to the different water and gas adsorption capacities of the IL [56], which affect the initial TGA values and the silica Si–OH degradation process, the real IL content in the hybrids could not be calculated based on the initial and final values of the TGA run.
Eq. 1
Calculation of IL mass percentage in silica xerogel, where m %A —mass at the beginning of degradation, m %B —mass at the end of the degradation, obtained from superimposed DTG/TGA curves; and \(m_{{\bmod \cdot {\text{SiO}}_{2} }}\)—initial mass of modified silica
Eq. 2
Calculation of IL theoretical content in silica xerogel, where n IL/n TEOS = 0.03, \(M_{{{\text{SiO}}_{2} }}\) = 60 g/mol, and M IL corresponds to the molecular weight of each particular IL
When IL were confined into the silica networks, these presented broader T d ranges and a general decrease in thermal resistances, compared to the neat IL [57]. This reduced thermal stability is a probable consequence of the higher surface area presented by the small IL domains dispersed on the silica surface [58]. The [BF4] anion-based samples presented lower thermal resistances, which could be due to HF formation [47]. For this anion, the T d of the IL with shorter alkyl cation side-chain was higher than with the longer one. Those with the [NTf2] anion presented higher T d values, following the behavior observed for neat IL. Additionally, all silicas with [NTf2] anion-based IL presented a two-step degradation. It is possible that, in the first degradation stage of this confined IL, carbon species have been formed [59]. Thus, the formation of a carbon “layer” would delay the IL’s further decomposition and cause a second degradation stage shifted to higher temperatures.
After each TGA (under N2 atmosphere), the silica ashes were collected to give an insight into the diversity of decomposition mechanisms possible depending on the IL confined into silica (Fig. 8). Most of the residual ashes present highly distinct coloration, which appears to be related to the chemical composition of the IL applied into the process. The coloration intensity for some silica xerogels (Fig. 8c, e, f) is coincident with higher (up to 2 %) char residue of pure IL (Fig. 7a curves 5 and 6). Curiously, all [NTf2]-based IL presented intense coloration, even the most thermal stable ones. Most interestingly, this phenomenon seems not to be related with a thermo-oxidative process as none of the samples applied to calcination under air atmosphere presented coloration. This behavior indicates the occurrence of a distinct [NTf2]-based IL decomposition process under N2 atmosphere in a confined environment through the formation of silica–carbon species [59].

Silica xerogel residual ashes obtained from TGA (900 °C, at N2 atmosphere): a C4MImBF4, b C10MImBF4, c C7O3MImBF4, d C4MImNTf2, e C10MImNTf2 and f C7O3MImNTf2
4 Conclusions
The application of IL in the sol–gel process allowed a dynamic templating, from the first moments of the reaction, for obtaining silica with decreased random interparticle aggregation. This direct interaction with growing systems was confirmed by the presence of microporosity, fractal structuring, decrease in t gel and morphological changes. Samples with the [BF4] anion, a nucleophilic catalyst, presented a monomer–cluster growth mechanism that resulted in a dramatic decrease in the t gel and the presence of opalescence in both wet gels and xerogel nanoparticles. The capability of multiple H-bonding of ether-functionalized IL, as well as the different geometries and polarities of the applied anions, promoted the formation of “exotic” silica, e.g., corn grain or crescent-like shapes, which are even more defined when obtained by a casting technique. The understanding of the individual functions of each part of the IL’s molecular structure allows the formation of highly tuned nanosystems and nanoparticles solutions using very small contents of these multifunctional additives, which influence every step of the nanoparticles evolution process.
References
Pagliaro M, Ciriminna R, Palmisano G (2009) J Mater Chem 19:3116
Pénard AL, Gacoin T, Boilot JP (2007) Acc Chem Res 40:895
Nassif N, Livage J (2011) Chem Soc Rev 40:849
Radin S, Chen T, Ducheyne P (2009) Biomaterials 30:850
Karout A, Pierre AC (2007) J Non-Cryst Solids 353:2900
Klabunde KJ (2001) Nanoscale materials in chemistry. Wiley-Interscience, New York
Ciriminna R, Carà PD, Sciortino M, Pagliaro M (2011) Adv Synth Catal 353:677
Regí MV, Colilla M, González B (2011) Chem Soc Rev 40:5967
Ab Rahman I, Padavettan V (2012) J Nanomater 2012:1
Kim KD, Kim HT (2002) J Sol-Gel Sci Technol 25:183
Brinker CJ, Scherer GW (1990) Sol–gel science. Academic Press, New York
Letailleur LA, Ribot F, Boissière C, Teisseire J, Barthel E, Desmazières B, Chemin N, Sanchez C (2011) Chem Mater 23:5082
Curran MD, Gedris TE, Stiegman AE (1999) Chem Mater 11:1120
Estella J, Echeverría JC, Laguna M, Garrido JJ (2007) Microporous Mesoporous Mater 102:274
Poddenezhnyi EN, Kravchenko IP, Meľnichenko IM, Kapshai MN, Boiko AA (2000) Glass Ceram 57:158
Bruinsma PJ, Kim AY, Liu J, Baskaran S (1997) Chem Mater 9:2507
Nakamura H, Matsui Y (1995) J Am Chem Soc 117:2651
Yang J, Chen W, Ran X, Wang W, Fana J, Zhanga W (2014) RSC Adv 4:20069
Wasserscheid P, Welton T (2008) Ionic Liquids in Synthesis. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Schrekker HS, Donato RK, Fuentefria AM, Bergamo V, Luís B, Oliveira F, Machado MM (2013) Med Chem Commun 4:1457
Bergamo VZ, Donato RK, Dalla Lana DF, Donato KZ, Ortega GG, Schrekker HS, Fuentefria AM (2015) Lett Appl Microbiol 60:66
Riduan SN, Zhang Y (2013) Chem Soc Rev 42:9055
Le Bideau J, Viau L, Vioux A (2011) Chem Soc Rev 40:907
Tang S, Baker GA, Zhao H (2012) Chem Soc Rev 41:4030
Hallett JP, Welton T (2011) Chem Rev 111:3508
Xu C, Tang R, Hua Y, Zhang P (2008) Chin J Chem Phys 21:596
Dai S, Ju YH, Gao HJ, Lin JS, Pennycook SJ, Barnes CE (2000) Chem Commun 3:243
Vioux A, Viau L, Volland S, Le Bideau J (2010) C R Chim 13:242
Adams CJ, Bradley AE, Seddon KR (2001) Aust J Chem 54:679
Zhou Y, Antonietti M (2004) Chem Mater 16:544
Zhou Y, Antonietti M (2003) Chem Commun 20:2564
Zhou Y, Antonietti M (2003) Adv Mater 15:1452
Donato RK, Migliorini MV, Benvegnú MA, Stracke MP, Gelesky MA, Pavan FA, Schrekker CML, Benvenutti EV, Dupont J, Schrekker HS (2009) J Sol-Gel Sci Technol 49:71
Tang S, Liu S, Ren H, Liang X, Qiu H, Guo Y, Liu X, Jiang S (2014) RSC Adv 4:25819
Safaei S, Baltork IM, Khosropour AR, Moghadam M, Tangestaninej S, Mirkhani V (2013) Catal Sci Technol 3:2717
Donato RK, Benvegnú MA, Furlan LG, Mauler RS, Schrekker HS (2010) J Appl Polym Sci 116:304
Donato RK, Donato KZ, Schrekker HS, Matějka L (2012) J Mater Chem 22:9939
Donato RK, Matějka L, Schrekker HS, Pleštil J, Jigounov A, Brus J, Šlouf M (2011) J Mater Chem 21:13801
Schrekker HS, Silva DO, Gelesky MA, Stracke MP, Schrekker CML, Gonçalves RS, Dupont J (2008) J Braz Chem Soc 19:426
Migliorini MV, Donato RK, Benvegnú MA, Gonçalves RS, Schrekker HS (2008) J Sol-Gel Sci Technol 48:272
Ueno K, Imaizumi S, Hata K, Watanabe M (2009) Langmuir 25:825
Hench LL, West JK (1990) Chem Rev 90:33
Rahman IA, Vejayakumaran P, Sipaut CS, Ismail J, Bakar MA, Adnan R, Chee CK (2006) Ceram Int 32:691
Bergström L (2002) Handbook of applied surface and colloid chemistry. Wiley, New York
Donato RK, Lavorgna M, Musto P, Donato KZ, Jager A, Štěpánek P, Schrekker HS, Matějka L (2015) J Colloid Interface Sci 447:77
Jones RAL (2002) Soft condensed matter. OUP Oxford, Oxford
Freire MG, Neves CMSS, Marrucho IM, Coutinho JAP, Fernandes AM (2010) J Phys Chem A 114:3744
Dupont J, Spencer J (2004) Angew Chem Int 43:5296
Brinker CJ, Sehgal R, Hietala SL, Deshpande R, Smith DM, Loy D, Ashley CS (1994) J Membrane Sci 94:85
Kozuka H, Sakka S (1989) Chem Mater 1:398
Dupont J (2004) J Braz Chem Soc 15:341
Martinez JR, Palomares S, Zarzosa GO, Ruiz F, Chumakov Y (2006) Mater Lett 60:3526
Hea F, Hea X, Lib M, Zhangc S (2008) J Ceram Process Res 9:389
Mountjoy G, Rigden JS, Anderson R, Wallidge GW, Newport RJ, Smith ME (2000) J Mater Res 15:1998
Maton C, de Vos N, Stevens C (2013) Chem Soc Rev 42:5963
Deyko A, Jones RG (2012) Faraday Discuss 154:265
Gupta AK, Singh MP, Singh RK, Chandra S (2012) Dalton Trans 41:6263
Singh MP, Singh RK, Chandra S (2014) Prog Mater Sci 64:73
Wang X, Dai S (2010) Angew Chem Int Ed 49:6664
Acknowledgments
The authors thank the Brazilian funding agencies CAPES, CNPq and FAPERGS for financial support, as well as CNR-Italy and AVCR—Czech Republic for funding the cooperation project “New sustainable approaches in the synthesis of epoxy–silica hybrids with tunable properties (2013–2015).” L. Matějka gratefully appreciates the financial support of the Grant Agency of the Czech Republic (P108/12/1459). R.K. Donato is thankful to FAPERGS-CAPES for the DOCFIX postdoctoral fellowship. The authors are thankful to Gustavo P. Gonçalves for the AFM images.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Donato, K.Z., Donato, R.K., Lavorgna, M. et al. Ionic liquids as dynamic templating agents for sol–gel silica systems: synergistic anion and cation effect on the silica structured growth. J Sol-Gel Sci Technol 76, 414–427 (2015). https://doi.org/10.1007/s10971-015-3790-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-015-3790-5