
Abstract
Rapid radioanalytical methods are important in the case of a radiological emergency and for the defence against nuclear hazards, especially for pure alpha and beta emitters like 239/240Pu and 89Sr/90Sr. A new fast method was developed with an overall analysis time altogether around 11 h, for only strontium isotopes about 7 h. The method combines two extraction chromatography resins, DGA- and Sr-resin, to separate mainly strontium and plutonium. A broad variety of food samples with different fat, carbohydrate and protein contents were tested and successfully analysed. The yields obtained were typically around 95% and 70% for 90Sr and 242Pu.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the event of a radiological emergency the contamination of food products, especially with alpha and beta emitters, can be a tremendous hazard for the population. Therefore, the rapid determination is required in order to take effective countermeasures. Regarding pure alpha emitters the plutonium isotopes 238Pu and 239/240Pu are some of the most important isotopes in such an event due to their high dose coefficients (effective dose) for ingestion (e.g. 239/240Pu: 2.5E−07 Sv Bq−1 [1]). One of the most important pure beta emitters is radiostrontium, particular in regard of ingestion (dose coefficient for ingestion: 89Sr: 2.6E−09 Sv Bq−1, 90Sr: 2.8E−08 Sv Bq−1 [1]). Especially because of the similar chemical behaviour of strontium to calcium, leading to an enrichment of strontium in bones. However, the determination of the activity levels of pure alpha and pure beta emitters in food requires a complex radiochemical treatment, most of all because of self-absorption effects in volume samples. Another reason in the case of pure beta emitters is the continuous beta spectrum, which does not allow a high selectivity without an element-specific radiochemical treatment. In the case of pure alpha emitting radionuclides, the element-specific radiochemical treatment helps to minimize the self-absorption effects in the alpha source and the overlapping of the peaks in the spectrum. Therefore, the development of radioanalytical fast methods is highly required [2].
In this study the focus lies on food samples, which are different from other environmental samples like soil or groundwater. The main difference between them is the nature of the sample matrix and accordingly the sample pre-treatment step. In groundwater samples the focus lies more on the pre-concentration of the isotopes. For soil samples, the complete dissolution and matrix interferences are the most challenging part in the sample pre-treatment. Food samples contain a lot of carbohydrates, proteins and fat (so a plenty of organic material). The radiochemistry of an organic material differs significantly from that of a mainly inorganic material like groundwater or soil sample. This aspect should be considered in the sample pre-treatment. By food samples the focus is on the destruction of organic matter with the goal to apply afterwards classical radioanalytic [3].
The most important radiostrontium isotopes are the two fission products 89Sr (T1/2 = 50.56 days) and 90Sr (T1/2 = 28.79 years). A recent review gives a good overview of the available methods to determine radiostrontium in environmental samples [4].
A common approach for the fast determination of radiostrontium is co-precipitation with oxalate, then rapid column separation with strontium resin cartridges, followed by liquid scintillation counting (LSC) [5]. The Sr-resin uses a dicyclohexano-18-crown-6 derivate as extractant, which retains strontium under acidic conditions. Nowadays new resin materials are under development, such as plastic scintillation resins which are able to do both simultaneously, separation and detection of strontium [6]. Mass spectrometric methods (MS) to detect strontium are emerging, but they are still not an equivalent to beta measurements via LSC or low-level beta counting because of isobaric and polyatomic interferences and the comparatively high detection limits [7].
For the separation of plutonium isotopes from other actinides a wide range of column materials can be used, including anion exchange resins and extraction chromatographic resins like TEVA-resin or DGA-resin [8,9,10]. The DGA-resin uses the N,N,N′,N′-n-octyldiglycoamide as extractant. It shows a high affinity to Pu and americium in nitric- and hydrochloric medium [2]. α-spectrometry allows the determination of 238Pu and 239/240Pu. However, the determination of the ratio of 239Pu to 240Pu is impossible due to overlapping peaks. The plutonium source for the α-measurement can be prepared by electrodeposition or by microcoprecipitation. As radioactive tracer 242Pu or 236Pu can be used. Another way to measure long-lived plutonium isotopes is via Inductively Coupled Plasma (ICP)–MS [11]. ICP–MS has a short measurement time and allows the determination of the isotopic ratio 239Pu/240Pu, since it measures atomic masses. In the case of a low activity level of 238Pu and consequently a low mass of 238Pu, interferences with uranium arise. Furthermore, the ICP-MS method is inadequate to determine 238Pu due to its relatively short half-life [12]. Moreover, highly contaminated samples cannot be analysed with an ultra-sensitive mass spectrometric instrument without diluting the samples. In the case of a sample with unknown activity levels of plutonium isotopes, α-spectrometric measurements are indispensable. Therefore, ICP-MS measurements can complement α-spectrometry in some cases, but cannot replace it.
The aim of the present study was to develop a fast method to determine the activity concentrations of 90Sr and plutonium isotopes in food samples. In this study, the Sr-resin was chosen to separate radiostrontium, because it is a well-established material for the separation of strontium [2] and has been proven successfully in the development of other fast methods [6, 13]. To separate plutonium the DGA-resin was chosen as column material because of its promising features in separating not only plutonium, but also other actinides, which is an important feature in respect of further developments of this method [2]. The simultaneous separation of plutonium isotopes and radiostrontium isotopes in the same sample aliquot has the advantage to be faster than two separate procedures and smaller sample volumes are needed.
New fast methods to detect radiostrontium and plutonium isotopes, which can provide faster information about the contamination of food are crucial in an emergency case and can have a great impact of the health of the population.
Materials and methods
Chemicals
All chemical reagents and Atomic Absorption Spectroscopy (AAS) standards were obtained from Merck Chemicals, Germany. The chemicals were of analytical grade. The calcium and barium AAS standards were Certipur® standard solutions and the strontium AAS standard was a Certipur® ICP standard. The 2 mL Sr- and the DGA-cartridges were obtained from Triskem International, France. The 90Sr tracer was obtained from Physikalisch-Technische Bundesanstalt (PTB), Germany, the 241Pu tracer was obtained from AEA Technology, UK, and the 242Pu tracer was obtained from National Institute of Standards and Technology (NIST), USA. The scintillation cocktail Ultima Gold AB was obtained from Perkin Elmer, USA.
Samples
In order to cover a wide range of different carbohydrate, fat and protein contents, a broad variety of food samples, like salad, cereals, hazelnut spread, grapes and full cream milk were analyzed. The fat, carbohydrate and protein content of each food matrix tested in the experiments is listed in Table 1.
All food samples, except for the wild boar meat and the pasta casserole, were bought in a local supermarket. The wild boar meat was obtained from a hunter from Garmisch-Partenkirchen, Germany. The pasta casserole was homemade.
Equipment
For microwave digestions a Mars 6 instrument from CEM, Germany, was utilized. Beta measurements were performed by LSC, using an ultra-low-level Wallac Quantulus 1220 system from Perkin Elmer, USA. Elementary analysis was performed by AAS with a contrAA 700 spectrometer from Analytik Jena, Germany. Alpha measurements were performed using a PIPS detector in a Canberra Alpha Analyst alpha spectrometric system from Canberra, Germany. For the radiochemical separation a polycarbonate vacuum box system from Triskem International, France, was used.
Experimental
Sample pretreatment
Four different approaches were tested to prepare the samples: an open wet acidic digestion, an acidic microwave digestion, ashing the sample in a microwave muffle furnace and ashing the sample in a muffle furnace with subsequent dissolution. These four approaches were tested with three different matrices: salad, cereals and hazelnut spread.
The open wet acidic digestion was tested with 11 g of sample, 15 mL of concentrated nitric acid (65%) and 6 mL hydrogen peroxide (30%). The sample was slowly heated up to 130 °C on a hot plate.
The two ashing methods were tested with 20 g of sample per vessel with different programmes varying the heating steps and time from 2 h till 5 h until the end temperature of 600 °C was reached.
Furthermore, the acidic microwave digestion was tested with different concentrated nitric acid (65%)–hydrogen peroxide (30%) ratios (5:2 to 5:4), with different amounts of sample per microwave vessel (from 0.3 to 1 g) and different temperatures (150–180 °C) using time programs from 5 to 25 min excluding the cooling time.
Preliminary separation tests
Prior to the experiments with spiked food samples some preliminary tests were done. The separation of the three alkaline earth elements calcium, strontium and barium was tested in water on the 2 mL Sr-cartridges with carriers and at different concentrations of nitric acid (2–8 M).
The 2 mL DGA-cartdridge was tested with a 6 M HNO3 loading solution containing 0.5 mg calcium carrier, 241Pu and 242Pu tracer solutions. Then the cartridge was rinsed with 6 M HNO3. Two different elution schemes were applied.
Separation scheme I
Calcium was removed from the cartridge with 8 mL of deionized water. Plutonium was eluted with 20 mL 0.5 M HCl–0.1 M NH2OH solution. An aliquot of 5 mL from the plutonium fraction was taken and mixed with 15 mL of the Ultima Gold AB cocktail for an LSC measurement of pure beta emitter 241Pu. Then the plutonium source for the alpha-measurement was prepared by microcoprecipitation according to the alpha measurement description below for the determination of alpha-plutonium.
Separation scheme II
Calcium was removed from the cartridge with 8 mL of a 0.1 M NaNO2 solution. Americium (the decay product of 241Pu) was eluted with 10 mL of a 0.5 M HCl–0.1 M NaNO2 solution. Finally, plutonium was eluted with 10 mL of a 0.1 M NH4HC2O4 solution.
Moreover the optimal way to stack the Sr and the DGA-resin was examined.
Experimental set-up for food samples
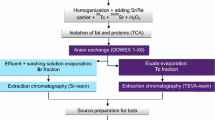
The procedure to simultaneously determine alpha and beta emitters, like plutonium and strontium, in food samples is shown in Fig. 1. First, the food samples were prepared for the experiments. Solid food was cut in small pieces and milk was stirred before processing. For solid food samples the use of a meat grinder or other homogenizer was intentionally omitted to avoid cross contaminations. In order to get homogeneous samples in the case of the solid food samples special care was taken to include all parts, like for example for the wild boar meat, the fatty and the muscle parts. Approximately 11 g of solid food and 55 mL of milk, respectively, were taken for the experiments. 0.5 mg of each strontium, calcium and barium carriers were added to the samples in order to monitor the chemical yield and the separation of the three alkaline earth metals. Additionally, the samples were spiked with 90Sr, 241Pu and 242Pu tracers. Next, the samples were dissolved via microwave digestion with a cocktail consisting of 5 mL of concentrated nitric acid (65%) and 2 mL of hydrogen peroxide (30%). After digestion, the solution was evaporated to dryness and the residue was dissolved in 10 mL 6 M nitric acid. The appearance of the solution was examined.

Separation scheme of the fast method to determine radiostrontium and plutonium isotopes in food samples
The oxidation state of plutonium was adjusted in two steps. First, 1.5 mL hydrazinium hydroxide was added to reduce plutonium to Pu(III). In the second step, Pu(III) was oxidized with 1 g NaNO2 to Pu(IV). Depending on the fat and protein content of the sample, a filtration step using a 0.45 µm cellulose nitrate filter was needed before the separation on the cartridges. In this arrangement on Fig. 1 a DGA-cartridge was stacked on a Sr-cartridge and placed on the vacuum box. All the fractions were collected in plastic LSC vials. From these fractions an aliquot was taken for the AAS measurement. The flow rate was 1 mL min−1 for the elution steps and 3 mL min−1 for the rinse steps. The cartridges were preconditioned with 6 mL of 6 M nitric acid before loading the sample solution. After loading the sample solution, the cartridges were rinsed with 6 mL of 6 M nitric acid and separated. In order to save time the elutions of strontium and plutonium from the cartridges were done simultaneously.
From the Sr-cartridge barium was eluted with 8 mL of 8 M nitric acid and afterwards strontium was eluted with 8 mL of deionized H2O. The separation scheme II was applied to separate calcium, americium and plutonium on the DGA-cartridge.
α-Spectrometric and LSC measurement
The strontium fraction was mixed with 15 mL of the Ultima Gold AB cocktail and measured for 1 h in a Quantulus 1220 instrument. The α-source was prepared by microcoprecipitation with neodymium fluoride. 50 µL of a 1 mg mL−1 neodymium nitrate solution and 1 mL of conc. hydrofluoric acid were added to the plutonium fraction. After 15 min the solution was filtered through an 0.1 µm polypropylene filter and the filter was washed twice with 1.5 mL of deionized water each and finally with 1.5 mL of ethanol. The filter was dried for 45 min. Then, the filter was counted for 3 h by the alpha spectrometer. Details on the alpha-spectra are given in the next chapter.
Results and discussion
Sample pretreatment
Four approaches were tested and several aspects were taken into account to select the most suitable methods for an emergency situation: the time needed for a complete dissolution of the sample, the amount of residue, the colour and visual appearance after the dissolution, the safety aspect for the laboratory staff, the sample quantity and the goal to find one general approach for all types of food samples.
The open wet acidic digestion took over 12 h to digest the food samples. The acidic microwave digestion dissolved the food samples within 1 h. The other two methods, ashing the sample in a microwave muffle furnace and ashing the sample in a muffle furnace, both took over 5 h to ash the samples. In total about 130 runs were done to identify the optimal sample preparation procedure suitable for any kind of food sample. With regard to the criteria mentioned above the acidic microwave digestion was selected as the most suitable pretreatment method.
In order to be as time efficient as possible, the microwave digestion was optimized to be as fast as possible with the highest possible amount of sample per vessel. After evaluating the experiments, the maximum amount was set to 1 g of solid food per vessel and 5 mL of liquid samples per vessel, totalizing 11 g of solid food and 55 mL of liquid samples per run, respectively. A digestion time of 20 min and a temperature of 180 °C was found to be the most reliable and efficient program to digest the samples. For hazelnut spread a special setup was found as optimum.
Preliminary separation test results
The first experiments had the goal to optimize the separation of the alkaline-earth elements barium, calcium and strontium. They were performed using barium, calcium and strontium carriers and a strontium tracer (90Sr) dissolved in deionized water. Different nitric acid concentrations (2–8 M) were tested on the Sr-resin to achieve the separation of strontium from barium and calcium. It was found that a load with 6 M nitric acid, a rinse step with 6 M nitric acid and a Ba removal step with 8 M nitric acid was the optimum to achieve the best separation of all three elements from each other.
Based on the optimized separation procedure the two different ways to stack the Sr- and the DGA-resin on top of each other were tested. When Sr-cartridge was stacked on DGA-cartridge the separation of barium from strontium and from calcium and plutonium was not sufficient. When the DGA-cartridge was stacked on the Sr-cartridge a clean strontium and a clean plutonium fraction was obtained. So the further experiments were done with the second setup, the DGA-cartridge stacked on the Sr-cartridge.
After the separation of plutonium applying the separation scheme I, the alpha spectrum of the plutonium fraction showed a 241Am peak. 241Am (T1/2 = 432 a) is the daughter of 241Pu (T1/2 = 14.33 a) [2]. This was due to the fact that a 241Pu tracer solution was employed for the determination of the effectiveness of the microcoprecipitation step. 241Pu can be measured by LSC quite fast so it is an ideal tracer to monitor the efficiency of the microcoprecipitation step. 241Am decays to 237Np emitting alpha particles at energies about 5.48 MeV and 5.44 MeV. These emissions were seen in the Pu alpha spectrum. Before the microcoprecipitation an aliquot of the plutonium sample was measured by LSC to compare afterwards the yield of 242Pu with the yield of 241Pu. A loss of activity from 10 to 20% due to the microcoprecipitation was observed. If required, the method can be applied also for the measurement of pure beta emitting 241Pu in the samples. In this case only 242Pu or 236Pu should be added as tracer.
In order to separate americium from plutonium, the separation scheme II was developed. After the calcium removal, americium was stripped with 10 mL of 0.5 M HCl solution. This resulted in a small leakage of plutonium in the americium fraction. To achieve a clean separation, plutonium was stabilized with NaNO2 by changing the calcium removal solution from deionized water to a 0.01 M NaNO2 solution and the americium strip solution to a 0.5 M HCl–0.1 M NaNO2 solution. Furthermore, the plutonium strip solution was changed to a 0.1 M NH4HC2O4 solution, because test experiments showed that the volume of the strip solution can be reduced to 10 mL with this adjustment.
Food experiments
The optimized method was then applied to a variety of food matrices to ensure that the method is suitable for any food matrix.
All samples were fully digested without any residue after the microwave digestion. After evaporating the solution to dryness the samples show a residue. This residue dissolved for the most of the samples completely in the 6 M nitric acid.
The first experiments were performed on matrices like salad, wild boar meat and grapes. In the next step food samples (cereals, pasta casserole, milk) with higher fat, carbohydrate and protein contents were tested. Samples with higher fat contents required a filtration step after the adjustment of the oxidation state of plutonium, leading to a minimal time delay of about 15 min. From all the matrices analysed until now, the hazelnut spread was the most complex one. The preparation of this matrix took the most time and the problems faced by this matrix were the most challenging to resolve due to the high fat and carbohydrate content. For this sample the microwave digestion parameters had to be slightly changed due to the unexpected high pressure formed in the microwave vessels during digestion. The temperature was set to 150 °C and the digestion time was increased by 5 min.
The LSC-spectra were processed with the spectral deconvolution technique. This technique allows the determination of both strontium isotopes 89Sr and 90Sr including also the SQP dependence, therefore the SQP values were not separately specified.
The results of the experiments on spiked food samples are summarized in Table 2. The yields of 90Sr and 242Pu were typically around 95% and 70%, respectively. Compared to other results found in literature [6, 14] a near quantitative yield for strontium can be considered as a good result. The lower yield of plutonium in comparison to strontium is due to some additional loss during the microcoprecipitation.
Possible interferences
Beside the americium issue, which was successfully resolved by adapting the separation scheme II, in a few cases the alpha- spectra of Pu showed some traces of natural thorium and radium isotopes as can be seen in Fig. 2. Some of the α-spectra of plutonium showed thorium and radium peaks (black) in addition to the 242Pu peak (red). These peaks are traces of the natural uranium and thorium series. This could be an issue, because the thorium and radium peaks can interfere with the 238Pu peaks and 239/240Pu peaks [13]. Further work has to be done to separate plutonium from traces of thorium and radium, which may be eluted from the DGA-resin together with the plutonium fraction using this method.

α-Spectrum of the plutonium fraction separated from the hazelnut spread sample
Time requirement
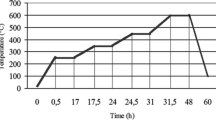
One of the main project objectives was to perform the analyses as quickly as possible. For the radiochemical separation, a vacuum box with cartridges was applied with a flow rate of 1 mL min−1. The time interval needed for each step is presented in Fig. 3. The simultaneous determination of strontium and plutonium radioisotopes can be realized in an emergency situation within 11 h, thus within a day. The results for strontium were obtained even faster, after 7 h, because the Sr eluate can be measured directly with LSC for 1 h after the radiochemical separation. The plutonium fraction has to be microcoprecipitated after the radiochemical separation and is then measured for 3 h by alpha spectrometry.

Analysis time needed for the determination of radiostrontium and plutonium isotopes
Quality control
The fast method was tested with a national intercomparison sample of raw milk for strontium (Milk 3), which was simultaneously analyzed with the standard strontium method in our laboratory (Milk 1 and Milk 2) [14]. In this experiment the activities of both 89Sr and 90Sr were determined by LSC measurement. The results are shown in Table 3. The results of the activity concentrations of 89Sr and 90Sr are in good agreement with the values obtained with the standard strontium method. Unfortunately, the target value of the intercomparison exercise has not been published yet, so an evaluation of the method was not possible.
To ensure that the method is generally applicable to food, the most complex matrix among the investigated food samples, hazelnut spread, was tested for repeatability. The results are presented in Table 4. The mean yield and the standard deviation were 91.6 ± 6.3% for 90Sr and 81.7 ± 7.7% for 242Pu.
The quality control of the radiostrontium analysis was supported by AAS measurements. The stable isotopes of strontium, calcium and barium were measured with AAS to monitor the chemical yield of strontium and the separation of the three alkaline earth metals. The obtained results for the other samples were similar in regard of the separation of strontium, barium and calcium The AAS results of the wild boar meat experiment are shown in Fig. 4 as an example. The largest part of barium and calcium is not retained on the columns, but found in the effluent and the first washing fraction. A small fraction of barium and calcium is retained on the Sr-resin and eluted with the barium fraction. A part of the calcium is retained on the DGA-resin and eluted with the calcium fraction. No calcium or barium is found in the strontium or plutonium fraction. All the strontium is found in the strontium fraction.

Results of the AAS measurement of the wild boar meat experiment
Uncertainty analysis and detection limits
The uncertainty analysis and the determination of the detection limits were performed with the software UncertRadio 2 version 2.1.8 2017/07 according to ISO 11029 [15]. The detection limit of 90Sr was 0.5 Bq kg−1 for solid food and 0.1 Bq L−1 for milk. The combined overall uncertainty for the activity concentration of 90Sr was around 15% with a coverage factor of k = 2. In the case of the uncertainty analysis of plutonium, the detection limit of 239/240Pu was 0.1 Bq kg−1. The overall uncertainty for the activity concentration of 239/240Pu will be determined in the future using an adequate standard reference material because the samples analysed during the method development does not contain any 239/240Pu.
Conclusions
A fast method based on extraction chromatography was developed for the determination of radiostrontium and plutonium isotopes in food samples. A DGA- and a Sr-resin cartridge were combined to separate strontium and plutonium. The chemical yields obtained were typically around 95% and 70% for 90Sr and 242Pu, respectively. The overall analysis time for the method is around 11 h. If only radiostrontium has to be determined, the overall analysis time reduces to 7 h. The method is applicable also for determination of 241Pu. The detection limits of strontium are about 0.5 Bq kg−1 for solid food samples and 0.1 Bq L−1 for milk with one hour counting time. The detection limit of 239/240Pu was 0.1 Bq kg−1 with 3 h counting time. It was demonstrated that the method is suitable for a large variety of food samples. The method needs further work concerning the contamination of the plutonium α-source with natural radionuclides. In case of a radiological emergency this method is suitable due to its quickness and its sensitivity to give a fast response to the decision makers.
References
Delacroix D, Guerre JP, Leblanc P et al (2002) Radionuclide and radiation protection data handbook. Nuclear Technology Publishing, Ashford
Lehto J, Hou X (2011) Chemistry and analysis of radionuclides. Wiley-VCH, Weinheim
Maxwell SL (2008) Rapid method for the determination of plutonium, americium and curium in large soil samples. J Radioanal Nucl Chem 275:395–402
Shao Y, Yang G, Tazoe H et al (2018) A review of measurement methodologies and their applications to environmental (90)Sr. J Environ Radioact 192:321–333
Guerin N, Riopel R, Rao R et al (2017) An improved method for the rapid determination of 90Sr in cow’s milk. J Environ Radioact 175–176:115–119
Sáez-Muñoz M, Bagán H, Tarancón A et al (2018) Rapid method for radiostrontium determination in milk in emergency situations using PS resin. J Radioanal Nucl Chem 315:543–555
Habibi A, Cariou N, Boulet B et al (2017) Automated chromatographic separation coupled on-line to ICP-MS measurements for the quantification of actinides and radiostrontium in soil samples. J Radioanal Nucl Chem 314:127–139
Maxwell SL, Culligan B, Hutchison JB et al (2018) Rapid method to determine plutonium, neptunium, americium and curium in granite samples. Appl Radiat Isot 140:102–108
Groska J, Vajda N, Molnár Z et al (2016) Determination of actinides in radioactive waste after separation on a single DGA resin column. J Radioanal Nucl Chem 309:1145–1158
Shin C, Choi H, Kwon HM et al (2017) Determination of plutonium isotopes (238, 239, 240Pu) and strontium (90Sr) in seafood using alpha spectrometry and liquid scintillation spectrometry. J Environ Radioact 177:151–157
Wang Z, Wen W, Quan W et al (2018) Rapid determination of Pu isotopes for decommission concrete samples by inductively coupled plasma mass spectrometry. J Radioanal Nucl Chem 316:411–417
Vajda N, Kim CK (2011) Determination of transuranium isotopes (Pu, Np, Am) by radiometric techniques: a review of analytical methodology. Anal Chem 83:4688–4719
Worsfold P, Townshend A, Poole C, Miró M (eds) (2019) Encyclopedia of analytical science, 3rd edn. Elsevier, Amsterdam
Kabai E, Hornung L, Savkin BT et al (2011) Fast method and ultra fast screening for determination of 90Sr in milk and dairy products. Sci Total Environ 410–411:235–240
Kanisch G (2016) Generalized evaluation of environmental radioactivity measurements with UncertRadio. Part I: methods without linear unfolding. Appl Radiat Isot 110:28–41
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dolique, I., Kabai, E. & Schuster, M. Fast method for the determination of radiostrontium and plutonium isotopes in food samples. J Radioanal Nucl Chem 322, 1423–1430 (2019). https://doi.org/10.1007/s10967-019-06850-w
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-019-06850-w