
Abstract
A new method was developed to effectively separate Am(III) from Cm(III). Am(III) was selectively oxidized to Am(V) using a mixture of Na2S2O8, Ag(I), and NaOCl in 0.01 M HNO3. Cm(III) was selectively retained on a DGA resin, while Am(V) had no retention. A separation factor of 110 ± 20 was usually obtained from a single separation. The new separation method was applied to determine Cm isotopes 244, 245 and 246 by accelerator mass spectrometry (AMS) in spent nuclear fuel samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The measurement of Am and Cm in irradiated nuclear materials is of interest for nuclear waste management [1], spent nuclear fuel recycling [2,3,4,5,6,7], nuclear fuel analysis, and nuclear forensics [8]. The measurement of some isotopes of Am or Cm by alpha or mass spectrometry can be challenging. For example, the isotopes 243Cm and 244Cm have energies too similar to be resolved by alpha spectrometry (5883 and 5902 keV, respectively for the main peaks [9]), therefore mass spectrometry is required to measure the individual isotopes. However, 243Am cannot be resolved from 243Cm with mass spectrometry techniques such as thermal ionization mass spectrometry (TIMS), inductively coupled plasma mass spectrometry (ICP-MS), or compact accelerator mass spectrometry (AMS). Thus, to properly quantify Am and Cm isotopes in the same sample, the optimal solution is to effectively separate these 2 elements. Also, in irradiated nuclear materials, there is often significantly more Am than Cm, which could result in undesirable contamination of the ion source of the mass spectrometer with Am when measuring Cm isotopes. It could make it more difficult subsequently to measure trace amounts of Am isotopes if desired. In the cases mentioned, it would be advantageous to separate Am from Cm; but, the separation of Am(III) and Cm(III) is extremely challenging due to their very similar chemistry in aqueous solution. Current methods to separate Am and Cm are tedious and usually result in low separation factors (SF) [5, 10]. More efficient separation methods are needed.
The main Am isotopes (241Am and 243Am) are commonly measured by alpha spectrometry as they have relatively short radioactive half-lives (t1/2): 432.6 ± 0.6 a [9] and 7367 ± 23 a [9] for 241Am and 243Am, respectively. Cm isotopes are measured by radiometric or mass spectrometry methods. Alpha spectrometry can be used to measure 243Cm (t1/2 = 28.9 ± 0.4 a [9]) plus 244Cm (t1/2 = 18.11 ± 0.03 a [9]) at trace levels, but it is not usually sufficiently sensitive to measure the lower abundant high mass Cm isotopes such as 245Cm (t1/2 = 8250 ± 70 a [9]), 246Cm (t1/2 = 4723 ± 27 a [9]), and 247Cm (t1/2 = 1.56 × 107 a [9]). Long-lived Cm isotopes are usually measured by mass spectrometry techniques such as TIMS [11], ICP-MS [12], and more recently by low energy compact AMS [13, 14].
The chemistries of tri-valent Am and Cm in aqueous solutions are strikingly similar making them difficult to separate [15, 16]. Normally, Am in acidic aqueous solution is of oxidation state (III) [17], but under rare select conditions in aqueous solutions, Am(III) can be oxidized to Am(V) or Am(VI) [18]. However, Cm(III) is much more difficult to oxidize. After selectively oxidizing Am(III) to higher oxidation states, Am can be separated from Cm(III) by precipitation or ion exchange chromatography [19, 20]. The preferred strategy used to separate Am from Cm is through the selective oxidation of Am(III) to Am(V). Am(III) is first oxidized to a higher oxidation state (mixture of Am(V) and Am(VI)) using ammonium persulfate ((NH4)2S2O8) in dilute nitric acid (HNO3). Then, Am(VI) is reduced to Am(V) using sodium hypochlorite (NaClO) [1, 5, 7, 10, 18, 21]. Another strategy employed is to selectively oxidize Am(III) to Am(VI) using ((NH4)2S2O8) and silver nitrate (AgNO3) in a dilute HNO3 solution [2]. However, Am(VI) is unstable in nitric acid solutions and tends to reduce to Am(V) (and gradually to Am(III)) [18].
The optimal conditions to oxidize Am(III) to Am(V) or Am(VI) have been studied, but have not led to a simple and effective method to separate Am and Cm. Burns et al. [5] first suggested in 2012 that a column separation would be possible. Then, they demonstrated a partial separation using ion exchange chromatography, but low SF were obtained [20]. Mincher et al. [10] tried to separate Am(V) from Cm(III) in 2015 using a commercial TRU resin, but they observed that a significant amount of Am(V) was reduced to Am(III) by the resin (about 50%). They then tested inorganic materials to see if they could improve the separation. They calculated the distribution coefficient (Kd) of Am(V) and Cm(III) on these inorganic materials and predicted a SF of up to 142 after 24 h equilibrium.
An effective and simple method to separate Am and Cm is needed. This work describes such a method. Am(III) was oxidized to Am(VI) with persulfate and silver and then Am(VI) was reduced and stabilized to Am(V) using hypochlorite [22]. The Am(V) was stable for an extended period of time (> 3 days). Cm(III) was not oxidized in these conditions. Cm(III) was selectively retained on a DGA (Eichrom) resin but not Am(V). The separation between Am and Cm was optimized and tested on irradiated nuclear fuels to demonstrate the applicability of the developed method.
Experimental
Reagents and standards
Radiochemical isotope standard solutions of 241Am, 243Am, 244Cm were purchased from the National Institute of Standards and Technology (NIST), (Gaithersburg, MD, USA) and 248Cm from Oak Ridge National Laboratory (Oak Ridge, TN, USA). Trace metal grade nitric acid (HNO3) and hydrochloric acid (HCl) were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Extraction chromatography (EXC) resins UTEVA and DGA normal (50-100 μm) were purchased pre-packed in 2 mL cartridges from Eichrom Technologies, Inc. (Lisle, IL, USA). The main extracting agents of the UTEVA and DGA (normal) resins are dipentyl pentylphosphonate and N,N,N’,N’-tetra-n-octyldiglycolamide, respectively. De-ionized water used for this work was obtained from a Millipore Direct-Q5 purification system (Billericia, MA, USA). Sodium persulfate (Na2S2O8), silver nitrate (AgNO3), and 10-15% sodium hypochlorite (NaClO) solution were obtained from Sigma-Aldrich (Oakville, ON, Canada).
Equipment
An Octete Plus® Alpha Spectrometer with eight 450 mm2 ULTRA-AS ion-implanted silicon detectors (AMETEK/ORTEC Inc., Oak Ridge, TN, USA) was used for method development tests. A coaxial high purity germanium (HPGe) detector with 10 cm of lead shielding (AMETEK/ORTEC Inc., Oak Ridge, TN, USA) was used to identify the fission products present in the fuel samples and to verify that Am was properly removed before preparing the AMS targets. The compact 0.6 MV AMS system TANDY (ETH Zurich, Zurich, Switzerland) was used to measure separated Cm isotopes [23].
Separation procedure
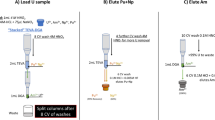
The procedure has two parts: Am(III) oxidation and DGA column separation as shown in Fig. 1.

Flow chart of the separation method
The sample, containing Am(III) and Cm(III), was evaporated to dryness in a 20 mL glass vial. Note, if the sample is initially in hydrochloric acid solution, convert to nitrate form by evaporation twice with concentrated nitric acid. A volume of 10 mL of 0.16 M Na2S2O8 + 0.005 M AgNO3 in 0.01 M HNO3 was added to the residue and the vial was heated for 20 min at 80 °C using a water bath (Fig. 1, steps 1 and 2). Note that a suspension of Ag2O is formed in the reagent solution and the solution shall be shaken before being used. The sample was taken out of the water bath and 1 mL of 33 mM NaOCl reagent was immediately added to the solution (Fig. 1, step 3). A white precipitate of AgCl was formed. Immediately after the addition of NaOCl, the solution acidity was adjusted to ~ 1 M HNO3 by adding 1.5 ml of 8 M HNO3 (Fig. 1, step 4). The solution was left to cool to room temperature, transfered to a centrifugation container, and centrifuged (Fig. 1, step 5).
The DGA resins were mounted on top of a 12-hole vacuum box. The 2 mL pre-packed columns were conditioned by passing 10 ml of 1 M HNO3. The supernatant was passed through the DGA resin at a flow rate of about 1 mL min−1 to selectively extract Cm(III) (Fig. 1, step 6). The resin was rinsed with 10 ml of 1 M HNO3 (Fig. 1, step 7). The load solution plus rinse were collected for Am analysis if necessary as the Am(V) is not retained by the resin under these conditions [24]. Cm(III) was eluted from the resin using 15 mL of 0.1 M HCl (Fig. 1, step 8).
Method development
The formation of Am(V) was optimized by studying the effect of the concentration of three reagents: Na2S2O8, HNO3 and AgNO3. The concentration of Na2S2O8 was varied from 0.08 to 0.42 M in 0.01 M HNO3 without the use of AgNO3. The Na2S2O8 concentration was then held at 0.16 M and two concentrations of AgNO3, which were in large excess from Am, were evaluated (0.005 and 0.01 M). Finally, nitric acid concentrations were varied from 0.01 to 2 M, with 0.16 M Na2S2O8 and 0.005 M AgNO3. An excess of NaClO was used, thus was not further optimized.
The stability of Am(V) was evaluated utilizing 243Am, with measurement by alpha spectrometry. A solution was prepared as per the procedure section. Aliquots of the solution were taken periodically up to 14 days and Am(V) and (III) were separated using a DGA resin. All alpha sources were prepared using the CeF3 micro-precipitation described by Dai [25].
The SF of the method was determined by using about 100 mBq of 241Am or 244Cm. The test samples were passed through all the steps of the method described in the separation procedure above. The sample activities were measured by alpha spectrometry and then the SF was calculated.
The SF is defined for this work as the activity (or mass) ratio of a given element before and after chemical separation from another element in a given fraction or sample. For example, if a sample initially contains 100 mBq of Am and 200 mBq of Cm and after separation, 1 mBq of Am is measured in this fraction, the SF in this example would be 100 (100 mBq/1 mBq).
The typical activities of 241Am or 244Cm used for the method development tests were between 15 and 30 mBq.
Application of the method to nuclear fuel samples
This method was tested on irradiated nuclear fuels originating at the Chalk River Laboratories (Chalk River, ON, Canada). These fuel samples had high levels of Am relative to Cm, making the separation desirable before measurement by AMS. These samples are highly radioactive due to the presence of fission and activation products. To assist with safety in handling, an initial separation was undertaken to remove the majority of the uranium, plutonium and fission products.
The fuel was dissolved in a nitric acid solution in a hotcell. A small fraction of the resultant solution was removed from the hotcell and made to 8 M HNO3. A known amount of 248Cm tracer (~ 10 pg) was added to the samples for Cm recovery calculations. The solutions were passed through UTEVA and DGA stacked columns. After separation, the columns were dismantled and Am, Cm and Ln were eluted from the DGA column using 20 mL of 0.1 M HCl. An aliquot of 2 mL of the eluate was evaporated to dryness in a small Teflon beaker. The residue was dissolved with 5 mL of 16 M HNO3 and evaporated again to dryness to ensure removal of HCl in the sample. The spent nuclear fuel samples were then processed according to the Am/Cm separation procedure described above. If the procedure needs to be repeated to obtain a higher SF, the elution fraction from the DGA resin (15 ml of 0.1 M HCl, as given in Fig. 1, step 8) was evaporated to dryness. The residue obtained was then converted to nitrate form by evaporation twice with concentrated nitric acid. Then, the residue was re-dissolved with 10 mL of 0.16 M Na2S2O8 + 0.005 M AgNO3 in 0.01 M HNO3, which enabled to repeat the Am/Cm separation procedure. The purified Cm was prepared for AMS measurement according to Dai et al. [13]. Americium-241 was measured by gamma spectroscopy before and after separation to estimate the method SF.
Radioactivity reduction
Following the preliminary separation steps described above (UTEVA + DGA resins), significant radioactivity remained in some high burnup samples as shown in Fig. 2a. Gamma spectroscopy analyses showed the radioactivity originated from Eu isotopes in the solution. To reduce the radioactivity, it was chosen to separate lanthanides (III) from actinides (III) on a TEVA resin using Eichrom method [26]. Briefly, starting with evaporation of the DGA resin eluate (Fig. 1, step 8), dissolve the actinides and lanthanides in a solution of 4 M KSCN + 0.1 M formic acid. Pass the resultant solution through a TEVA resin column, which selectively retains the actinides in these conditions but not the lanthanides. Finally, elute the actinides from the TEVA resin using a 1 M HCl solution. After Ln removal, no Eu was detected by gamma spectroscopy and the Am/Cm was fully recovered (Fig. 2b).

Gamma spectrum of the purified fuel solution. a Before and b after removal of the lanthanides
Results and discussion
Optimization of the formation of Am(V)
By adding a small amount (0.08 M) of persulfate to a 0.01 M nitric acid solution containing initially Am(III) and Cm(III), most of the Am was not retained on the DGA column (~ 80%) while almost all of the Cm was retained, as shown in Table 1. Increasing the amount of persulfate from 0.08 to 0.42 M did not significantly improve the oxidation from Am(III) to Am(V) (Table 1). Since the pre-loading step in this column work included the addition of hypochlorite, which is known to reduce Am(VI) to Am(V) [22], it suggests that a portion of the Am was not converted to Am(V) or Am(VI). A concentration of 0.16 M Na2S2O8 was chosen for subsequent experiments.
A concentration of silver nitrate of 0.005 and 0.01 M, in addition to the persulfate, significantly reduced the retention of Am on the DGA resin to 1.1 ± 0.7 and 1.2 ± 0.4%, respectively (see Table 2) compared to about 15% without AgNO3. The presence of silver promoted the oxidation of Am to higher oxidation states. As the results were similar for both concentrations tested, 0.005 M of silver was chosen for subsequent work.
The final reagent concentration optimized for this work was nitric acid. By increasing the concentration of nitric acid from 0.01 to 2 M, Am was less retained on the DGA resin as shown in Table 3. This is in agreement with published results [7, 10, 22]. Am(V) is less favored when the nitric acid concentration increases. As per the experimental section, 0.01 M HNO3 was chosen as the optimum concentration.
The overall effect of the reagents is summarized in Fig. 3 illustrating the sucessful separation of Am and Cm.

Optimization summary of the Am and Cm separation on DGA resin in various conditions: a 0.01 M HNO3, b 0.16 M Na2S2O8 in 0.01 M HNO3, and c Mix of 0.16 M Na2S2O8 + 0.005 M AgNO3 in 0.01 M HNO3
The elution fraction of the DGA resin (Fig. 1 step 8) should contain less Am after separation, which was the case since a SF of 110 ± 20 for Am was obtained (Table 4). The absence of Cm in the load and rinse solution fractions eluted from the DGA column (Fig. 1 steps 6 and 7) was also verified by obtaining a SF of ≥ 3300 ± 200 for Cm (Table 4). This is expected given that Cm(III) is known to be strongly retained on the DGA resin. A separation factor of about 110 for Am in the resin elution fraction is comparable to similar methods [10]. The separation method is rapid, therefore the Cm fraction can be purified several times if needed to obtain a higher SF. This is the strategy used to purify Cm from Am in the application section described below.
Stability of Am(V) species
It is crucial to maintain Am as Am(V) in solution long enough to be able to separate it from Cm(III) using a DGA resin. The stability of Am(V) in solution was tested for 14 days (Fig. 4). Americium(V) was slowly reduced over time as demonstrated by increased retention of Am on the DGA resin (Fig. 4). Importantly, once converted to Am(V), the oxidation state remained almost unchanged for at least 3 days, (variation of Am extraction < 2%). Subsequently, Am(V) very slowly reduced to Am(III) (Fig. 4). This is comparable to previously published work [5]. However, Am(V) was not significantly reduced by the DGA resin as it has been observered with the TRU resin [10].

Percentage of Am extracted on the DGA resin as a function of time to test Am(V) stability
Application to irradiated nuclear fuel
The measurement results for selected irradiated fuel samples are shown in Table 5. The Am SF was around 90 ± 20 for samples 1–5 for one separation (Table 5). For sample 6, a higher SF was desired to reduce the amount of Am lower or close to 1 pg for AMS measurement; therefore, a second separation was performed. A much higher SF was obtained (> 1030 ± 70). This Am/Cm separation factor is significantly higher than previously published work [5, 10]. The time for a single separation is less than 2 h, thus this is a rapid and convenient method. Should a high SF be required, it is quick to repeat. For interest, the amount of 244Cm, 245Cm, and 246Cm measured in the samples are presented in Table 5. To the best of our knowledge, this is the first measurement of Cm isotopes in spent nuclear fuel sample by AMS.
For samples 1, 2, 4, and 6, the 241Am activity measured by gamma spectrometry after separation was equal to the background. For these samples the activity measured after separation was estimated as the minimum activity that could be detected and the value was reported with the sign less than (see Table 5). The SF calculated from these values are reported with the sign greater than since the real SF value is superior to what is reported, but cannot be calculated more precisely (see Table 5). It explains why after a double separation a SF > 1030 ± 70 was obtained when a value of approximately 8100 would be expected (90 × 90).
Conclusion
A new effective method was developed to selectively separate Am from Cm. An effective separation was obtained by optimizing the conversion of Am(III) to Am(V) and maintaining this oxidation state for a long time (> 3 days). Am(V) was not reduced by the DGA resin. A sample batch can be processed in just a few hours providing rapid turnaround. A single separation gave a SF of 110 ± 20. This rapid and easy separation method can be repeated to obtain much higher SF if desired. The new developed method has potential applications in nuclear fuel analysis, radio-chronometry, forensics and environmental studies.
References
Dares CJ, Lapides AM, Mincher BJ, Meyer TJ (2015) Electrochemical oxidation of 243Am(III) in nitric acid by a terpyridyl-derivatized electrode. Science 350:652–655
Kamoshida M, Fukasawa T (1996) Solvent Extraction of Americium(VI) by Tri-n-Butyl Phosphate. J Nucl Sci Technol 33(5):403–408
Chapron S, Marie C, Pacary V, Duchesne M-T, Arrachart G, Pellet- Rostaing S, Miguirditchian M (2016) Separation of Americium by Liquid-Liquid Extraction Using Diglycolamides Water-Soluble Complexing Agents. Procedia Chem 21:133–139
Geist A, Taylor R, Ekberg C, Guilbaud P, Modolo G, Bourg S (2016) The SACSESS hydrometallurgy domain—an overview. Proc Chem 21:218–222
Burns JD, Shehee TC, Clearfield A, Hobbs DT (2012) Separation of americium from curium by oxidation and ion exchange. Anal Chem 84:6930–6932
Modolo G, Geist A, Miguirditchian M (2015) Reprocessing and recycling of spent. Nucl Fuel Chap. https://doi.org/10.1016/B978-1-78242-212-9.00010-1
Burns JD, Moyer BA (2016) Group hexavalent actinide separations: a new approach to used nuclear fuel recycling. Inorg Chem 55:8913–8919
Whitney SM, Biegalski S, Buchholz B (2007) Analyzing nuclear fuel cycles from isotopic ratios of waste products applicable to measurement by accelerator mass spectrometry. Nucl Sci Eng 157:200–209
Recommended data, Laboratoire National Henri Becquerel, http://www.nucleide.org/DDEP_WG/DDEPdata.htm. Accessd Feb 2018
Mincher BJ, Schmitt NC, Schuetz BK, Shehee TC, Hobbs DT (2015) Recent advances in f-element separations based on a new method for the production of pentavalent americium in acidic solution. RSC Adv 34(5):27205–27210
Poupard D, Journaux B (1990) Determination of picogram quantities of americium and curium by thermal ionization mass spectrometry (TIMS). Radiochim Acta 49:25–28
Chartier F, Aubert M, Pilier M (1999) Determination of Am and Cm in spent nuclear fuels by isotope dilution inductively coupled plasma mass spectrometry and isotope dilution thermal ionization mass spectrometry after separation by high-performance liquid chromatography. Fresenius J Anal Chem 364(4):320–327
Dai X, Christl M, Kramer-Tremblay S, Hans-Arno S (2016) Determination of Atto- to Femtogram Levels of Americium and Curium Isotopes in Large-Volume Urine Samples by Compact Accelerator Mass Spectrometry. Anal Chem 88:2832–2837
Christl M, Dai X, Lachner J, Kramer-Tremblay S, Hans-Arno S (2014) Low energy AMS of americium and curium. Nucl Instrum Meth B 331:225–232
Myasoedov BF, Maryutina TA, Litvina MN, Malikov DA, Kulyako YuM, Spivakov BY, Hill C, Adnet J-M, Lecomte M, Madic C (2005) Americium(III)/curium(III) separation by countercurrent chromatography using malonamide extractants. Radiochim Acta 93:9–15
Šťastná K, John J, Ŝebesta F, Vlk M (2015) Separation of curium from americium using composite sorbents and complexing agent solutions. J Radioanal Nucl Chem 304:349–355
Schweitzer GK, Pesterfield LL (2010) The aqueous chemistry of the elements. Oxford University Press, New York
Runde WH, Mincher BJ (2011) Higher oxidation states of americium: preparation, characterization and use for separations. Chem Rev 111:5723–5741
Glenn AB (1968) Separation of americium from curium by precipitation of K3Am O2(CO3)2. Nucl Appl 4:217–221
Burns JD, Shehee TC, Clearfield A, Hobbs DT (2012) Separation of americium from curium by oxidation and ion exchange. Anal Chem 84:6930–6932
Richards JM, Sudowe R (2016) Separation of americium in high oxidation states from curium utilizing sodium bismuthate. Anal Chem 88:4605–4608
Burns JD, Borkowski M, Clearfield A, Reed DT (2012) Separation of oxidized americium from lanthanides by use of pillared metal(IV) phosphate-phosphonate hybrid materials. Radiochim Acta 100:901–906
Christl M, Vockenhuber C, Kubik PW, Wacker L, Lachner J, Alfimov V, Synal HA (2013) The ETH Zurich AMS facilities: performance parameters and reference materials. Nucl Instrum Meth B 294:29–38
Horwitz EP, McAlister DR, Bond AH, Barrans RE (2005) Novel extraction of chromatographic resins based on tetraalkyldiglycolamides: characterization and potential applications. Solvent Extra Ion Exch 23(3):319–344
Dai X (2011) Isotopic uranium analysis in urine samples by alpha spectrometry. J Radioanal Nucl Chem 289(2):595–600
Separation of Am from rare earths, Eichrom methods (2014) http://www.eichrom.com/eichrom/radiochem/methods/eichrom/index.aspx. Access Feb 2018
Acknowledgements
This research was undertaken for Atomic Energy of Canada limited and funded by the Canadian Federal Science and Technology program for Project 51200.50.18.06.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kazi, Z., Guérin, N., Christl, M. et al. Effective separation of Am(III) and Cm(III) using a DGA resin via the selective oxidation of Am(III) to Am(V). J Radioanal Nucl Chem 321, 227–233 (2019). https://doi.org/10.1007/s10967-019-06571-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-019-06571-0