
Abstract
A crown-ether-based extraction chromatography resin, Eichrom Pb resin, was characterized for separations of flerovium (Fl) homologs, specifically Pb and Sn. The batch uptake of Pb(II) and Sn(IV) radionuclides was determined from an HCl matrix. Both Pb(II) and Sn(IV) are strongly retained on the resin at different HCl concentrations. The affinity for Pb(II) decreases with increasing HCl concentration while Sn(IV) uptake increases. Extraction kinetics for Pb(II) and Sn(IV) were examined and show suitable uptake on the second time scale. Separation methods for the isolation of individual homologs, Pb(II) and Sn(IV), have been established using 2 mL pre-packed vacuum flow Pb resin columns.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chemical investigations of the transactinide elements (Z ≥ 104) present many challenges. The short half-lives and small cross sections at the nano- or picobarn levels result in low production rates, which means that transactinides need to be studied one atom-at-a-time. As a result, an individual atom is unable to interact with another atom of the same element, and can only interact with its surroundings. Therefore, studies of the homologs of the transactinides (elements in the same chemical group) must be carried out either on-line, by producing individual atoms at an accelerator and performing rapid chemical extractions immediately after their production, or with carrier-free, ultratrace radionuclides. To obtain reliable results it is necessary that the chemical system chosen allows the single atom to undergo many exchanges with the extractant molecule so that it mimics an equilibrium state over the short time-scale of the experiment [1]. Extraction chromatography gives the selectivity of a liquid–liquid extraction system with the ease of running a column, along with the benefits of a large number of metal–ligand interactions [2].
Reaction kinetics are important in the selection of a chemical system. The short half-life of Fl (289Fl, t1/2 = 1.9 +0.7−0.4 s [3]) means that the chemical system will never reach a true equilibrium state. Therefore, the atom of interest must undergo enough exchanges so that it reaches a pseudo-equilibrium state allowing for extrapolation of its chemical properties from the behavior of its homolog elements from the same chemical group in the same system. Each chemical system must be tested to ensure that it can operate on the time scales encountered during an on-line experiment (typically seconds to minutes). Initial studies presented here focus on finding an extractant with suitable kinetics for an on-line Fl chemistry experiment, with possible implementation once an automated chemical system that is capable of being run on the second time scale has been established.
The primary motivation for the study of transactinide elements is to investigate the role of relativistic effects on the chemical properties within a group of the Periodic Table. These effects lead to vastly differing predictions between the established trends of the lighter homologs within a group and the corresponding transactinide elements [4, 5]. Based on classical predictions, Fl appears in Group 14 of the Periodic Table and its chemistry should be similar to that of Pb and Sn. Considering the trend in Groups 13–17 of the Periodic Table, where as the atomic number Z increases down a group the elements attain more of a metallic character, Fl should therefore be more metallic than its nearest homolog, Pb. Relativistic calculations on Fl suggest an increased stability of the ground state, compared to that of Pb, with a configuration of [Rn] 5f146d107s27p 21/2 [6]. Some predictions have indicated a higher volatility and inertness leading to Fl behaving somewhat similarly to Hg or a noble gas [7–10]. Other more recent predictions expect Fl to be more volatile than Pb but still metallic in character [11]. Recent gas phase experiments have been performed on Fl with differing results as to its behavior [12, 13]. In order to investigate these deviations, the chemical properties of the transactinides must be compared to the properties of their lighter homologs. Therefore, extractants of interest for future chemical studies must have high selectivity for the homologs of Fl as well.
Crown ethers are macrocyclic ligands that exhibit highly selective extraction behavior for various metal atoms. The coordination chemistry of crown ethers has been studied in a variety of separations with a large number of different metals [14–16]. The high selectivity of crown ethers is attributed to their extraction ability, which is based on their charge density and ionic radius as determined by the ring size. In solution, the free crown ether has flexibility in its cavity size, but this can be inhibited by adding steric hinderances to the sides of the crown [15–17].
For the development of a suitable chemical system for Fl homologs, a commercially available extraction chromatography resin was evaluated for the separation of Pb and Sn. The resin, Eichrom Pb resin (50–100 µm particle diameter), is coated with 4′,4″(5″)-di-tert-butyldicyclohexano-18-crown-6 (DtBuC18C6) (Fig. 1), which as mentioned above separates analytes based on their size as well as their charge and complexation [14, 15, 17, 18]. This resin was developed for the specific application of separating Pb from other analytes; however, no research into the behavior of Sn has been performed previously.

Pb resin extractant 4′,4″(5″)-di-tert-butyldicyclohexano-18-crown-6 (DtBuC18C6)
Batch experiments were conducted to determine the extraction efficiency of the Pb resin for both Pb(II) and Sn(IV) from HCl. In acidic solution, Pb tends to stay in the +2 oxidation state, while Sn is stable as the +4 [19–21]. The extraction kinetics were investigated, and column separation schemes were developed for the separation of Pb(II) and Sn(IV) with different elution orders. To assess the speciation of Pb(II) and Sn(IV), liquid–liquid extraction experiments were performed with varying DtBuC18C6 concentrations. The primary focus of this work was to establish a suitable separation scheme for Fl, that gives direct insight into the chemical form of the extracted homologs with appropriate kinetics for potential future application to a Fl chemistry experiment.
Experimental
Reagents and materials
The Pb resin (50–100 µm, 40 % w:w, Eichrom Industries, Inc.) was used for both batch and column studies [18]. The extractant 4′,4″(5″)-di-tert-butyldicyclohexano-18-crown-6 (90 %) was purchased from Sigma Aldrich and used in liquid–liquid extraction experiments as received. Dichloromethane (99.9 %, un-stabilized, Fisher) was used without further purification. Acids were prepared from trace-metal grade acids and de-ionized water (18 MΩ cm). The tracer solutions of 212Pb and 113Sn were prepared with activity concentrations ranging from 2 to 10 cps per 20 µL. The 232U (legacy material, Lawrence Livermore National Laboratory (LLNL)) decay chain was used to obtain 212Pb as a tracer for all studies by milking the 212Pb from a generator [22]. The 113Sn tracer was produced at the Center for Accelerator Mass Spectrometry (CAMS) at LLNL via the natIn(p,n)113Sn reaction. The natIn foil (99.9 %, Goodfellow Inc.) was dissolved in 1 M HCl and the 113Sn was separated by passing the solution through an AG 1 × 8 anion exchange column (removing In) and eluting 113Sn with 3 M HNO3 as in Refs. [23, 24].
Activity measurements
Activity measurements were performed on a high purity germanium (HPGe) gamma-ray spectrometer coupled with a multi-channel analyzer (DSPEC, Ortec). The detector efficiency was between 0.05 and 11 % for the 75–1600 keV energy range. Spectral files were analyzed with Maestro spectral software (Ortec). The spectral lines with the highest relative yield were chosen for determining the activity of each radionuclide (Table 1) [25].
All post-extraction 113Sn counting was performed 24 h after conclusion of the experiment to allow the 113In daughter (which the activity is derived from) to reach secular equilibrium.
Batch uptake experiments
The uptake parameters for Pb(II) and Sn(IV) on Pb resin in HCl solutions were determined by batch extraction experiments. To a 1.5 mL centrifuge vial, 10–20 mg Pb resin were added along with 1 mL of HCl ranging in concentration from 0.001 M to concentrated. The resin was placed on a rotary mixer for 1 h to precondition the resin. A 20 µL spike containing either 212Pb (eluted from the generator with 2.0 M HCl) or 113Sn (oxidized to Sn(IV) with a drop of H2O2) in 2.0 M HCl was added to the wet resin. The solutions were equilibrated for 3 h on a rotary mixer, each sample was counted with a HPGe detector for 120–900 s (≥1000 counts under the desired photo-peak), and then filtered through a 0.45 µm polytetrafluoroethylene (PTFE) filter to completely separate the resin from the solution. A 700 µL aliquot of each filtered solution was added to 320 µL de-ionized water in a 1.5 mL centrifuge vial (to maintain initial counting geometry) and counted for 300–900 s (depending on activity) with the same HPGe detector used in initial counts. All experiments were performed in triplicate and the reported errors are based on the standard deviation of the replicates. The Pb resin capacity factor (k′), or the number of free column volumes to peak maximum, can be inferred from the batch extraction distribution ratio using the method described in the references [18, 26].
Uptake kinetics
The HCl concentration at which the maximum uptake (4 M HCl for 113Sn(IV) and 1 M HCl for 212Pb(II)) occurs in the batch experiments was the concentration of choice for kinetics studies. The standards were prepared by placing 1 mL of HCl at the desired concentration (depending on whether 212Pb or 113Sn was used) in a 1.5 mL centrifuge tube and adding 100 µL of a stock radionuclide solution. The standards were made in triplicate and counted with a HPGe detector for 120 s in the case of 212Pb and 3600 s in the case of 113Sn. The preconditioned samples were prepared (in triplicate) by adding 1 mL of the desired concentration of HCl to a 1.5 mL centrifuge vial containing 10–20 mg Pb resin and placing on a rotary mixer for 1 h. A 100 µL spike of either 212Pb(II) or 113Sn(IV) in the above mentioned HCl concentration was added to the samples, and each sample was mixed for a specific time interval before quickly filtering to isolate the solution from the resin. A 700 µL spike of each filtered solution was added to 400 µL of de-ionized water (to maintain original counting geometry) and the samples were counted with a HPGe detector for 240–300 s (212Pb) or 6300 s (113Sn).
Column experiments
The column extraction of mixed 212Pb(II) and 113Sn(IV) was performed with pre-packed 2 mL cartridges containing dry Pb resin. Aliquots of each tracer were combined and evaporated to dryness in a warm water bath with a forced air stream, then reconstituted in 1 mL of the appropriate HCl solution. The initial sample activity was determined by HPGe counting. For the extraction experiments, a 24-hole polycarbonate vacuum box (Eichrom, Darien, IL, USA) with a pressure regulator was used to accelerate the elution process to maintain an eluent flow rate of ~2 mL/min (~4 mmHg gauge reading). The resin cartridge was conditioned with 10 bed volumes (20 mL) of the appropriate HCl solution. Extractions were performed with HCl concentrations based on the results from the batch experiments. The radionuclides were loaded on the column in 3 M HCl, where both are retained, and 0.4 M HCl was used to elute 113Sn(IV) and 8 M HCl to elute 212Pb(II). Separate experiments were performed to reverse the elution order. Three rinse fractions at 3 M HCl were collected followed by 1 mL elution fractions (×10) of the desired HCl concentration. Care was taken to stop the column flow just as liquid was about to reach the top-frit so the column never ran dry and each elution fraction was a consistent 1 mL. Fractions were counted on a HPGe.
Speciation experiments
Solutions containing different concentrations of DtBuC18C6 in un-stabilized dichloromethane were prepared in volumetric flasks with volumes and masses as described for Pb and Sn speciation in Tables 2 and 3 respectively.
Stock solutions of 212Pb(II) were prepared by evaporating the generator eluted 212Pb(II) solution and reconstituting it in 0.4 M HCl (peak of the 212Pb(II) extraction from batch results). A 113Sn(IV) stock solution was prepared in 4 M HCl (peak of the 113Sn(IV) extraction from batch results). To a 1.5 mL centrifuge tube, 480 µL of either 0.4 or 4 M HCl was added along with 500 µL of crown ether solution (each DtBuC18C6 concentration was studied in triplicate for both Sn(IV) and Pb(II)). These were allowed to mix for one hour on a rotary mixer to ensure pre-conditioning of the organic phase. To each tube a 20 µL spike of the desired 212Pb(II) or 113Sn(IV) activity was added, and the phases were allowed to mix for one hour on a rotary mixer. A 300 µL aliquot from each phase was taken and counted with a HPGe detector to determine the distribution ratios for Sn(IV) and Pb(II) at each concentration of DtBuC18C6.
Results and discussion
Batch experiments
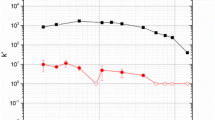
The effects of HCl concentration on the uptake of Sn(IV) and Pb(II) by the Pb resin are shown in Fig. 2. Lead shows a strong affinity to the resin from 0.04 to 2 M HCl and then it decreases significantly with increasing HCl concentration above 2 M, in good agreement with literature [18]. Tin shows a slight uptake from 0.04 to 1 M HCl and then the adsorption increases significantly to a peak extraction at around 4 M HCl. Currently there are no data available on the extraction of Sn(IV) with 18-crown-6 from hydrochloric acid media. It is expected that Sn(IV) in [HCl] > 0.7 M exists as the SnCl6 2− anion [20, 27, 28]. Crown ethers are known to form positively charged hydronium ion complexes by coordinating with H3O+, where the hydronium ion fits perfectly into the ring [29, 30]. Therefore, the increasing k′ for Sn(IV) above 1 M HCl suggests that hydronium activated crown ethers form an ion-association complex with the SnCl6 2− anion (this conclusion is drawn from data presented in Fig. 2 and in Fig. 5 below).

The batch uptake (k′) of 212Pb(II) and 113Sn(IV) as a function of hydrochloric acid media on Pb resin (50–100 μm) with a 3 h equilibration time. Error are from the standard deviation of replicates
Kinetics of Sn and Pb uptake on Pb Resin
The data obtained from the batch studies indicates that Sn(IV) can be separated from Pb(II) using a pure HCl matrix with the Pb resin at an equilibration time of three hours. The short-lived isotopes of Fl and the goal of an on-line chemical separation will require flow rates of mL min−1. Therefore, the kinetics of the extraction must be suitable on the second time-scale verses hours. The kinetics of Pb and Sn on the Pb resin at 1 and 4 M HCl (maximum k′ from batch studies), respectively, were investigated (Fig. 3a, b). The sorption of Pb(II) on the Pb resin was extremely fast with near immediate uptake and full equilibrium reached within five minutes. The sorption of Sn(IV) on the Pb resin was considerably slower than that of Pb(II), presumably from the participation of multiple crown ether/hydronium ion complexes (as discussed in Fig. 5) in the extraction of the negatively charged SnCl6 2− complex; however, significant extraction was seen in seconds, with full equilibrium was reached after approximately an hour.

a Kinetics of 212Pb(II) in 1 M HCl media and b 113Sn(IV) in 4 M HCl media on Pb resin (50–100 μm), varying equilibration times. Errors are from the standard deviation of replicates
Column experiments
The column experiments were used to determine if a stepwise extraction of Pb(II) and Sn(IV) could be achieved by varying only the HCl concentration. Based on the batch study results, a load solution of 3 M HCl was chosen due to the fact that both Pb(II) and Sn(IV) are retained on the Pb resin at this concentration. Two separate columns setups were run, one with the goal of removing Pb(II) before Sn(IV) and the other with the reverse order. To remove Pb(II) 8 M HCl was used while 0.4 M HCl was used to strip Sn(IV) (Fig. 4).

Column elution of 2 mL pre-packed Eichrom Pb resin cartridges at ~2 mL/min flow rate for the separation of Pb(II) from Sn(IV) with (top) Pb eluted first and (bottom) Sn eluted first. Errors presented are counting errors
Both analytes behaved as expected with virtually no breakthrough during column loading. Lead(II) was eluted completely with no Sn(IV) breakthrough and vice versa, usually within the first couple of free column volumes. These separation schemes are fast and yield an excellent separation between Pb(II) and Sn(IV) regardless of their elution order
Speciation experiments
Plotting the logarithm of the distribution ratios for 212Pb(II) and 113Sn(IV) as a function of the logarithm of the concentration of DtBuC18C6 yields a line where the slope of a linear fit to the line is equivalent to the number of DtBuC18C6 molecules required to extract each metal atom.
From the linear regressions in Fig. 5, the number of crown ligands coordinated to the Pb(II) metal ion is found to be 0.94 ± 0.02. This indicates that one DtBuC18C6 molecule is required to extract each Pb(II) ion into the organic phase, supporting the notion that Pb(II) extracts into the cavity of the crown ether. Similarly, the number of crown ligands coordinated to the Sn(IV) metal ion is found to be 1.61 ± 0.05. This suggests that a mixture of 1:1 and 1:2 (Sn(IV):DtBuC18C6) complexes are formed. This supports the idea that the highly stable SnCl6 2− complex is either being extracted by two positively charged DtBuC18C6·H3O+ complexes or one DtBuC18C6·H3O+ complex with charge balanced by an additional H3O+, leading to the ligand dependency value being less than 2.

Distribution ratios for the extracted 212Pb(II) and 113Sn(IV) as a function of DtBuC18C6 concentration in dichloromethane. The solid lines indicate the results of a linear regression fit to the Pb and Sn data, with slopes indicated
Conclusions
The extraction behavior of Pb(II) and Sn(IV) from HCl media was studied using Eichrom Pb resin, which contains the 4′,4″(5″)-di-tert-butyldicyclohexano-18-crown-6 extractant. In agreement with previously reported data, the batch results show Pb(II) extracts at low HCl concentrations, from 0.04 to 2 M. It was also observed that Sn(IV) extracts above 1 M HCl. The extracted Pb species is most likely the Pb2+ ion in the crown ether cavity, with the charge balanced by a Cl− counterion. Due to the formation of SnCl6 2−, Sn most likely extracts as an ion-association complex between the negatively charged Sn chloro-complex and positively charged hydronium crown ether complexes.
The results also showed that the reaction kinetics were relatively slow on the scale of minutes to hours to achieve full equilibrium. However, the k′ for both Pb(II) and Sn(IV) was >50 within a few seconds, indicating that the separation can be performed on the second timescale, even though complete equilibrium is not reached.
The column studies established separation schemes to isolate pure Pb(II) or Sn(IV) fractions from the Pb resin through the modification of the HCl concentration. The increased number of theoretical plates in the column system compared to that of the batch system allows for much faster flow rates, such as the 2 mL/min used in this work, while retaining full extraction of both analytes. Thus, the column experiments confirmed the results from batch studies and provide evidence that the Pb resin is suitable for the selective extraction of both Pb(II) and Sn(IV). If an appropriate apparatus was developed and the extraction behavior of Fl was studied using this same crown ether comparing to the behavior of Pb(II) and Sn(IV), the results would indicate whether Fl behaves more like Pb(II) or Sn(IV).
Speciation studies examined the species that was extracted by DtBuC18C6. The slope from linear regressions of the Kd of Sn(IV) and Pb(II) as a function of the concentration of DtBuC18C6 was found to be 1.61 ± 0.05 and 0.94 ± 0.02, respectively. This confirms the notion that Pb(II) extracts directly into the crown ether cavity, and gives strength to the notion that one (with H3O+ counterion) or two positively charged DtBuC18C6·H3O+ complexes extract one SnCl6 2− species.
Before an on-line Fl experiment can be performed, further experiments are necessary to determine the maximum rate at which the extraction can be carried out while maintaining the same level of separation. Future work is also needed to develop a continuous automated chemistry apparatus capable of running columns and preparing samples on the time scales required for a Fl experiment. Assuming an automated system was capable of performing the chemical separations and sample preparation on the desired time scales, the chemical system presented in this work would be capable of determining whether Fl in the aqueous phase is more Sn(IV) or Pb(II) like. An on-line experiment would first need to be performed and optimized with the short-lived Pb and Sn homologs produced in the same manner Fl would be, so direct comparisons between Fl and the homologs could be made. If Fl was seen in an on-line experiment optimized for Sn elution (high HCl concentrations) one would expect Fl to be forming more negatively charged complexes and be extracting based on ion exchange. Similarly, a system optimized for Pb elution (low HCl concentration) would indicate, if seen, that Fl extracts into the crown ether cavity most likely as a cation.
References
Guillaumont T, Adloff JP, Peneloux A (1989) Kinetic and thermodynamic aspects of tracer-scale and single atom chemistry. Radiochim Acta 46:169–176
Schädel M, Shaughnessy DA (eds) (2014) The chemistry of the super heavy elements. Springer, Heidelberg
YuTs Oganessian, Utyonkov VK (2015) Superheavy nuclei from 48Ca induced reactions. Nucl Phys A 944:62–98
Pershina V (1996) Electronic structure and properties of the transactinides and their compounds. Chem Rev 96:1977–2010
Pyykko P, Desclaux J-P (1979) Relativity and the periodic system of elements. Acc Chem Res 12:276–281
Pershina V (2010) Electronic structures and chemistry of the heaviest elements. In: Leszczynski J, Barysz M, Ishikawa Y (eds) Relativistic methods for Chemists. Springer, Dordrecht, pp 451–520
Pitzer KS (1975) Are elements 112, 114, and 118 relatively inert gases? J Chem Phys 63:1032–1033
Nash CS (2005) Atomic and molecular properties of elements 112, 114 and 118. J Phys Chem A 109:3493–3500
Liu W, van Wüllen C, Han YK, Choi YJ, Lee YS (2001) Spectroscopic constants of Pb and Eka-lead compounds: comparison of different approaches. Adv Quant Chem 39:325–355
Hoffman DC, Lee DM, Pershina V (2006) In: Vertes A, Klencsar Z (eds) The chemistry of the actinide and transactinide elements. Springer, Dordrecht, pp 1652–1752
Pershina V, Anton J, Fricke B (2007) Intermetallic compounds of the heaviest elements and their homologs: the electronic structure and bonding of MM′, where M = Ge, Sn, Pb and element 114, and M′ = Ni, Pd, Pt, Cu, Ag, Au, Sn, Pb, and element 114. J Chem Phys 127:134310
Eichler R, Aksenov NV, Albin YV, Belozerov AV, Bozhikov GA, Chepigin VI, Dmitriev SN, Dressler R, Gäggeler HW, Gorshikov VA, Henderson RA, Johnsen AM, Kenneally JM, Lebedev VY, Malyshev ON, Moody KJ, Oganessian YT, Petrushkin OV, Piguet D, Popeko AG, Rasmussen P, Serov A, Shaughnessy DA, Shishkin SV, Shutov AV, Stoyer MA, Stoyer NJ, Svirikhin AI, Tereshatov EE, Vostokin GK, Wegrzecki M, Wilk PA, Wittwer D, Yeremin AV (2010) Indication for a volatile element 114. Radiochim Acta 98:133–139
Yakushev A, Gates JM, Türler A, Schädel M, Düllmann CE, Eberhardt K, Essel HG, Even J, Forsberg U, Gorshkov A, Graeger R, Gregorich KE, Hartmann W, Herzberg R-D, Heßberger FP, Hild D, Hübner A, Jäger E, Khuyagbaatar J, Kindler B, Kratz JV, Krier J, Kurz N, Lommel B, Niewisch LJ, Nitsche H, Omtvedt JP, Parr E, Qin Z, Rudolph D, Runke J, Schausten B, Schimpf E, Semchenkov A, Steiner J, Thörle-Pospiech P, Uusitalo J, Wegrzecki M, Wiehl N (2014) Superheavy element flerovium (element 114) is a volatile metal. Inorg Chem 53:1624–1629
Yordanov AT, Roundhill MD (1998) Solution extractrion of transition and post-transition heavy and precious metals by chelate and macrocyclic ligands. Coord Chem Rev 170:93–124
Khopkar SM (2008) Analytical chemistry of macrocyclic and supramolecular compounds. Narosa Publishing House Pvt Ltd, New Delhi
Weber E, Toner JL, Goldberg I, Vögtle F, Laidler DA, Stoddart JF, Bartsch RA, Liotta CL (1989) Chapter 4 Crown ethers–complexes and selectivity. Crown Ethers and Analogs. Wiley, Chichester, pp 207–304
Izatt RM, Pawlak K, Bradshaw JS, Bruening RL (1991) Thermodynamic and kinetic data for macrocyclic interactions with cations and anions. Chem Rev 98:1721–2085
Horwitz EP, Dietz ML, Rhoads S, Felinto C, Gale NH, Houghton J (1994) A lead-selective extraction chromatographic resin and its application to the isolation of lead from geological samples. Anal Chim Acta 292:263–273
Seth M, Faegri K, Schwerdtfeger P (1998) The stability of the oxidation state +4 in group 14 compounds from carbon to element 114. Angew Chem Int Ed Engl 37:2493–2496
Nervik WE (1960) The radiochemistry of tin. Subcommittee on radiochemistry, National Acadamy of Sciences-National Research Council, Washington D.C
Gibson WM (1961) The radiochemistry of lead. Subcommittee on Radiochemistry, National Academy of Sciences-National Research Council, Washington D.C
Despotopulos JD (2015) Studies of flerovium and element 115 homologs with macrocyclic extractants (Doctoral dissertation). ProQuest, Ann Arbor 3715057:224
Rieman W, Walton HF (1970) Ion exchange in analytical chemistry. Pergamon, Oxford
Despotopulos JD, Kmak KN, Gharibyan N, Brown TA, Grant PM, Henderson RA, Moody KJ, Tumey SJ, Shaughnessy DA, Sudowe R (2015) Production and isolation of carrier-free homologs of flerovium and element 115 at the Lawrence Livermore National Laboratory Center for Accelerator Mass Spectrometry. J Radioanal Nucl Chem. doi:10.1007/s10967-015-4500-z
National Nuclear Data Center “NNDC”(2013) Brookhaven National Llaboratory, [Online]. Available: http://www.nndc.bnl.gov/. Accessed 16 Dec 2013
Horwitz EP, Bloomquist CA (1972) Preparation, performance, and factors affecting band spreading of high-efficiency extraction chromatographic columns for actinide separations. J Inorg Nucl Chem 34:3851–3871
Johnson JS, Kraus KA (1959) Hydrolitic behavior of metal ions IX. Ultracentrifugation of Sn(IV) in acidic chloride and perchlorate solutions. J Phys Chem 63:440–441
Kulrestha NK, Dey AK, Ghosh S (1955) Untersuchungen über hydratisiertes zinnoxyd. Kolloid-Z 141:106–109
Izatt RM, Haymore BL, Christensen JJ (1972) A stable OH3+–cyclic polyether complex characterised by infrared spectroscopy. J. Chem Soc Chem Comm 23:1308–1309
Junk PC (2001) Structural aspects of oxonium ion/crown ether complexes. Rev Anal Chem 21:93–124
Acknowledgments
The authors would like to thank the CAMS facility staff at LLNL, specifically Scott Tumey, Thomas Brown and Graham Bench for providing beam time and expertise to the production of radionuclides used in this study. This study was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. This work was funded by the Laboratory Directed Research and Development Program at LLNL under project tracking code 11-ERD-011, as well as by the LLNL Livermore Graduate Scholar Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Despotopulos, J.D., Kmak, K.N., Gharibyan, N. et al. Characterization of the homologs of flerovium with crown ether based extraction chromatography resins: studies in hydrochloric acid. J Radioanal Nucl Chem 310, 1201–1207 (2016). https://doi.org/10.1007/s10967-016-4917-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-016-4917-z