
Abstract
In order to evaluate the effect of the combination of micro-sized calcium carbonate (MCC) and nano-sized calcium carbonate (NCC) on the performance of poly(butylene adipate-co-terephthalate)/poly(lactic acid)/calcium carbonate (PBAT/PLA/CaCO3) composites by using a comparative approach, the composites were prepared by melt mixing. The melting and crystallization of the composites were investigated by differential scanning calorimetry. The non-isothermal crystallization kinetics was investigated by using Jeziorny and Mo methods. The mechanical properties of the composites were test and the fracture surfaces were observed by scanning electron microscopy. Nano and micron calcium carbonates (nano-CaCO3 and micro-CaCO3) together improved the heat resistance of the composites more effectively than them alone. Mo method indicated that the crystallization rate of the composites containing both nano-CaCO3 and micro-CaCO3 was between the nano-CaCO3 composites and the micro-CaCO3 composites. The composite containing both 5 parts nano-CaCO3 and 10 parts micro-CaCO3 had the highest tensile, yield and impact strengths among the composites, but the composite containing both 11 parts nano-CaCO3 and 5 parts micro-CaCO3 had the lowest. SEM displayed that the morphology of the dispersed PLA in the composites with both nano-CaCO3 and micro-CaCO3 was different from that in the composites with nano-CaCO3 and micro-CaCO3 alone. Controlling the ratio of nano-CaCO3 and micro-CaCO3 achieved the synergistic enhancement in PBAT/PLA/CaCO3 composites.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Most of the packaging materials currently used in supermarkets are non-biodegradable polyethylene, polypropylene, polyvinyl chloride and other resin-based disposable plastic bags. They bring great convenience in the huge consumption, but there are some difficulties in the recycling and reuse of these bags, more than 60% of which are buried or discarded, causing serious white pollution. To address the damage to the environment, there is a strong need to develop biodegradable and compostable packaging materials. Polybutylene terephthalate (PBAT) is a fully biodegradable polyester with excellent heat resistance, good ductility and toughness, making it very suitable as an alternative to polyethylene. However, its tensile modulus and tensile strength are lower than those of polyethylene and need to be modified [1,2,3]. In order to ensure the biodegradability of the product when modified, it is usually modified with another degradable resin, of which poly(lactic acid) (PLA) is the most commonly used. PLA is a biodegradable polyester obtained from starch, and has high modulus and strength comparable to many petroleum-based plastics [4,5,6]. However, the performance defects of PLA are also obvious, such as low melt strength, poor toughness and thermal stability [7]. PBAT and PLA have complementary properties, thus it seems that their co-blending modification is a good choice. With the modification, the PBAT/PLA blend can meet the performance requirements of packaging materials [8,9,10,11,12]. The main problem at present is that the cost of PBAT/PLA blends is higher than traditional non-degradable plastics, which limits their large-scale application in supermarkets. There are two main feasible ways to reduce the material cost, one is to add thermoplastic starch to PBAT/PLA blends [13,14,15,16] and the other is to add inorganic fillers (e.g. natural fibers [17, 18] and common minerals [19,20,21]). The combination of biodegradable blends with low-cost inorganic fillers not only reduces the cost but also improves the performance of the matrix. Among the common minerals, calcium carbonate (CaCO3) is widely used in combinations with PBAT/PLA blends [22, 23]. Although the wide range of applications of the combinations, there is a lack of in-depth research on the production of degradable materials, especially in the application of calcium carbonate in production.
There are two kinds of CaCO3 fillers commonly used, namely nano-sized and micro-sized [24,25,26]. Nano-sized CaCO3 can improve the rheological property and plasticity of plastic masterbatch in plastics processing, and can play a dual role of toughening and strengthening in plastics properties, so that the bending strength, elastic modulus, heat deflection temperature and dimensional stability of plastics can be improved, and thermal hysteresis can also be given to plastics. However, due to its large specific surface area and high surface energy, nano-sized CaCO3 is easily agglomerated, resulting in its enhanced properties not being realized. It would be too wasteful to use nano-sized CaCO3 only as a weight-adding agent. Compared to nano-sized CaCO3, micro-sized CaCO3 has received less attention. In fact, the surface of micro-sized CaCO3 particles is in a thermodynamically stable state, unlike nano-sized CaCO3, which is not easy to agglomerate and can achieve the performance of nano-sized CaCO3 in many plastics. The low production and use cost of micro-sized CaCO3 is favorable to further reduce the cost of biodegradable plastic bags and to increase their market share.
In order to evaluate the effect of micro-sized CaCO3 combining with nano-sized CaCO3 on the structure and properties of PBAT/PLA composite, the combination was investigated in the present study. The composites were prepared by melt-blending, and the performance of the composites was studied by using a comparative approach.
Experimental
Materials
PBAT (blow molding grade) was purchased from Lanshan Tunhe Co. Maleic anhydride grafted polylactic acid (LGM, G1603) was purchased from Foshan Zuogao Plastic Material Co. PLA (FY801, density: 1.24 g cm-3, MFR=4 g/10 min at 190 °C and 2.16 kg) was purchased from Anhui Fengyuan Co. Micro-sized CaCO3 (MCC) and nano-sized CaCO3 (NCC) were supplied by Nanzhao Dingcheng Calcium Industry Co. The characteristic parameters of CaCO3 fillers were listed in Table 1.
Preparation of PBAT/PLA/CaCO3 composites
The PBAT and PLA pellets were dried in a vacuum oven at 60 °C for 12 h before melt mixing to minimize the moisture. Afterward, PBAT and PLA with a mass ratio of 90/10 was fed into a mechanical mixer. In the case of PBAT/PLA/CaCO3 composites with micro- and nano-CaCO3, the fillers and LGM were added to the mixture of PBAT and PLA in the mixer. Table 2 provided the sample formulations and corresponding designations. Melt mixing was performed in twin-screw extruder at a screw speed of 60 rpm and temperatures ranging from 170 °C to 190 °C.
Differential scanning calorimetry
Thermal transitions of PBAT/PLA/CaCO3 composites were investigated using a differential scanning calorimeter (DSC) (DZ-DSC 300, Nanjing Dazhan Testing Instruments Co., Ltd.). The samples (9±1 mg) on DSC testing were obtained from the corresponding strips in the aforementioned tensile testing. The heating and cooling rates were both set at 20 °C/min. The samples were heated from 40 °C to 210 °C in a nitrogen atmosphere and held for 5 min, and then cooled to 40 °C. Afterwards, the cooled samples were reheated to 210 °C. The glass transition temperature (Tg), cold crystallization temperature (Tcc) and enthalpy of cold crystallization (∆Hcc), melting temperature (Tm) and enthalpy of melting (∆Hm) were obtained from the heating DSC curves. The degree of crystallization (Xc) was determined from the following Eq. (1).
where \(\Delta H_{m}^{0} = 144\) J g-1 for 100% crystalline PBAT [27] and \(w_{{{\text{PBAT}}}}\) is mass% of PBAT in the sample. The melt crystallization temperature (Tc) and crystallization enthalpy (∆Hc) were derived from the cooling DSC curves.
Non-isothermal crystallization kinetics
The samples were heated from 40 to 210 °C at 20 °C min-1 and held for 5 min. Subsequently, the samples were cooled to room temperature at -4, -6, -8 and -10 °C min-1, respectively. The crystallization time was converted by the following Eq. (2):
where t was the crystallization time, To was the initiate crystallization temperature, T was the temperature at time t, and φ was the cooling rate.
Tensile testing
The samples were tested by an electronic universal testing machine according to GB/T 1040.1-2006. Tensile rate was set at 20 mm/min. Repeated the test for 5 times for each sample and took the average value.
Scanning electron microscopy
The morphology of the fracture surfaces of the samples in the tensile testing was recorded by using a scanning electron microscope (SEM, Regulus8100, HITACHI, Japan) under a voltage of 5 kV at 5000 times magnification. The samples were gold coated before examination.
Wide angle X-ray diffraction
Wide angle X-ray diffraction (WAXD) diffraction patterns of the samples were recorded using an X-ray diffractometer (Rigaku SmartLab SE, Japan) using Cu-Kα radiation (λ = 0.154 nm). It was carried out in the reflection mode at 40 kV and 40 mA. Radial scans of intensity versus diffraction angle (2θ) were recorded in the range of 5° to 90° with a scanning rate of 10°/min and a step size of 0.02°.
Results and discussion
Melting and crystallization behaviors
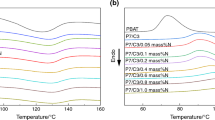
Li et al. [24] observed three distinct morphologies, i.e., spherical droplet, elongated fibrous structure, and co-continuous structure, in PLA/PBAT blend as the content of PBAT increased from 5 to 50 wt%. When the PBAT content reached 70 wt% and above, PLA droplets dispersing within the PBAT matrix. Deng et al. [8] presented a morphology diagram of the PLA/PBAT blend over a full range of composition, as deduced from the melt viscosity, optical micro graph, tensile properties and SEM fracture surfaces. PBAT and PLA formed co-continuous phase in the composition range between 19.0 and 40 wt% in the schematic diagram. When the PBAT content reached 40 wt% and above, PLA particles became dispersed in PBAT continuous matrix. When the PBAT content was more than 60 wt%, the size of the PLA particles became much finer and the dispersion became much more uniform, and the resulted blend was similar to PBAT. The PBAT-like thermal and mechanical properties of the composite still did not meet the requirements for use. Therefore, it was modified with CaCO3 filler in the present study. The PBAT/PLA blend with a weight ratio of 90 to 10 and its composites with nano- and micro-CaCO3 fillers were prepared. The melting and crystallization behaviors were investigated and the DSC thermograms were presented in Fig. 1.

DSC scans of PLA/PBAT/CaCO3 composites for both heating and cooling at 20 ℃/min. (A) , (C) and (E) were for first heating, cooling, and second heating, respectively, and (B), (D) and (F) were characteristic values derived from the DSC curves
As shown in Fig. 1, even if the PLA content was as low as 10 parts in the present study, its melting peaks were still obvious regardless of whether there was CaCO3 filler. PBAT had a very broad and shallow endotherm in the first heating indicating some crystallization of PBAT. In fact, PBAT is a random co-polymer and therefore dose not have a sufficiently symmetrical structure to give high levels of crystallinity [23]. However, the peak temperatures of melting endotherm and crystallization exotherm, whether attributed to PLA or PBAT, were affected by the adding content and particle size of the nano- and micro-sized CaCO3 fillers as shown in Fig. 1. In fact, CaCO3 particles affected the crystallization of PLA in the composites from two aspects, namely nucleation and cross-linking. CaCO3 particles had a strong nucleation effect and tended to promote PLA crystallization. However, the stronger cross-linking interaction with the matrix blocked the movement of molecular chain segments and reduced the crystallization rate and crystallinity of PLA in the PBAT/PLA/CaCO3 composites.
All the nascent PBAT/PLA/CaCO3 composites cooled naturally went through double melting in the first heating from 40 to 210 °C as shown in Fig. 1A. The melting at low temperature attributed to PBAT, and that at high temperature attributed to PLA. The diversity in DSC curves in Fig. 1A resulted from the addition of nano-CaCO3 and micro-CaCO3. Either the particle size or the content ratio of nano-CaCO3 and micro-CaCO3 had effect on the DSC curves. Compared with that in the Free-CC sample, the double melting temperatures in the 5NCC sample significant increased as shown in Fig. 1A. This showed that the presence of 5NCC not only made PBAT crystal more perfect, but also made PLA crystal more perfect. Actually, nano-CaCO3 was preferentially distributed in the PBAT phase, with a small amount in the PLA phase as shown in Fig. 2. Despite all this, a small amount of nano-CaCO3 significantly increased the melting temperature of PLA. Compared with that in the 5NCC sample, the double melting temperatures in the 11NCC sample decreased as shown in Fig. 1A. It was speculated that with the increase of nano-CaCO3 content, the trend of nano-CaCO3 aggregation increased, which was not conducive to the improvement of crystallization. Compared with that in the Free-CC and 11NCC samples, the double melting temperatures of the 5MCC and 11MCC samples increased, but still lower than that in the 5NCC sample. The diversity of DSC curves in the 5NCC/11MCC and 11NCC/5MCC samples was attributed to the synergies of nano-CaCO3 and micro-CaCO3.

Distribution and aggregation of CaCO3 particles in the composite
Figure 1C showed the -20 °C/min cooling DSC curves of PBAT/PLA/CaCO3 composites, PLA and PBAT. In literature [11, 28,29,30,31], the crystallization peak at high temperature was attributed the formation of the perfect crystallization of PLA, while that at low temperature was attributed to the imperfect crystallization of PLA. In this study, the high temperature exothermic peak on the DSC curve was reasonably attributed to PLA crystallization, and the low one to PBAT crystallization. For the Free-CC sample, there were two crystallization peaks in the cooling curve, indicating that both PBAT and PLA crystallized. But for the other composites containing the CaCO3 particles, there was only one crystallization peak at a low temperature, indicating that only PBAT crystallized. The absence of PLA crystallizing in PLA and PBAT/PLA/CaCO3 composites showed that the cooling rate at -20 °C/min was too fast to allow PLA to crystallize in time.
In fact, the CaCO3 particle played two roles in the crystallization of PLA and PBAT. One was that the CaCO3 particle acted as a heterophase nucleating agent in the crystalline matrix and shortened the crystallization induction time, defined as the promoting effect. The other was to form a strong connection with the matrix by van der Waals forces and reduce the activity of the matrix chains, defined as the retardation effect. Since there was not the CaCO3 particles, PLA and PBAT in the Free-CC sample crystallized. But the retardation effect in the composites containing the CaCO3 particle only allowed the crystallization of PBAT and not PLA crystallization. The higher crystallization temperature in the 5MCC sample was attributed to the higher promoting effect than that in other composites. In addition, the crystallization rate (i.e. Xc/t1/2) was also related to the CaCO3 particle. The high crystallization rate in the 5NCC and 5NCC/11MCC samples was due to the high number of heterogeneous nucleation active sites formed by the nano-CaCO3 particles at the level of 5 parts.
The DSC curves in the second heating were shown in Fig. 1c, and the derived parameters were listed in Fig. 1E-F. As shown, all samples in the study went through dual melting endotherms. The endotherm at high temperature, T’m, belonged to PLA melting, and the other, Tm, to PBAT melting. Tm and T’m of the 5NCC and 11NCC samples were both significantly higher than those of the Free-CC sample, where the 5NCC and 11NCC samples contained 5 and 11 parts of nano-CaCO3, respectively. In contrast, Tm and T’m of the 5MCC and 11MCC samples, which contained 5 and 11 parts of micro-CaCO3, respectively, were both slightly lower than those of the Free-CC sample. It seemed that the strong connection between nano-CaCO3 and the matrix in the 5NCC and 11NCC samples increased the wafer thickness of PLA and PBAT crystals and accordingly caused an increase in Tm and T’m, while the weak connection between micro-CaCO3 and the matrix in the 5MCC and 11MCC samples decreased the thickness and caused a decrease in Tm and T’m.
Non-isothermal crystallization of PLA/PBAT/CaCO3 composites
In order to further evaluate the effect of the cooling rate on the crystallization behavior of the composites, lower cooling rates than -20 ℃ min-1 were set, and the DSC thermograms were shown in Fig. 3. Two crystallization exotherms occurred in the DSC thermograms of all composites in the present study at the cooling rate of -4 ℃ min-1 as shown in Fig. 3a, and the peak temperatures were listed in Table 3. The exotherm around 90 ℃ was attributed to the formation of the imperfect crystalline of BAT, and the other around 125 ℃ to that of the perfect crystalline. In contrast, only one crystallization exotherm attributed to the imperfect crystalline occurred at -6, -8 and -10 ℃ min-1 except the Free-CC sample as shown in Fig. 3b-d. Regardless of the cooling rate, the Free-CC sample underwent double crystallization. It seemed that the presence of whether nano-CaCO3 or micro-CaCO3 reduced the mobility of PLA chains, so that the PLA chains only kept up with the rapid cooling rate at -4 ℃ min-1.

DSC scans of PBAT/PLA/CaCO3 on cooling at rates of (a)-4 ℃/min, (b) -6 ℃/min, (c) -8 ℃/min, and (d) -10 ℃/min
Xt versus time was shown in Fig. 4, and the characteristic parameters were listed in Table 4.. Here, Xt contained only the crystallinity of the imperfect crystalline. The half time of crystallization, t1/2, defined as the time required to reach 50% of the imperfect final crystallinity. As shown in Table 4., the 5NCC and 11NCC samples had a similar crystallization temperature (Tc), crystallinity (Xc) and crystallization rate (t1/2) at the same cooling rate, indicating that the increase of nano-CaCO3 had little effect on the crystallization of the composite. But, the 5MCC and 11MCC samples differed greatly in Tc, Xc and t1/2 at the same cooling rate. In fact, the 5MCC sample had the highest Tc among the composites in the present study, and had lower Xc and longer t1/2 than the 11MCC sample.

Plots of Xt versus t at cooling rates of -4, -6, -8, and -10 ℃/min for all the composites
Jeziorny method
The crystallization behavior at the low temperature was further investigated by the Jeziorny method. The isothermal crystallization kinetics is extensively described by the Avrami model [22], where Xt is expressed in the following form:
where n is the Avrami crystallization exponent, which depends on the nucleation mechanism and growth size; t is the crystallization time and Zt is the crystallization rate constant, which depends on nucleation and crystal growth. The above equation can be rewritten as a linear equation.
However, the Avrami model is not applicable to non-isothermal crystallization because the temperature constant changes and both nucleation and crystal growth are temperature dependent. Jeziorny proposed that Zt is influenced by the cooling rate (\(\varphi\)) and verified by having a cooling rate term as shown in equation (5) below [32].
where Zc is the corrected Jeziorny crystallization constant. According to Eq. (4), n and Zt in the present study were obtained from the slope and intercept of the curve between ln[-ln(1-Xt)] and lnt shown in Fig. 5. Then, the value of Zc was calculated by Eq. (3). n and Zc values of the composites were listed in Table 5.

Avrami plots for the Free-CC to 11NCC/5MCC samples
Compared with that of the Free-CC sample, the n value of the 5MCC sample was almost unchanged, indicating that the nucleation patterns in the Free-CC and 5MCC samples were close to each other. But the n value of the 11NCC sample increased significantly to 3.62, indicating that the pattern of PBAT crystal growth was three-dimensional. But the n value of the other samples increased slightly.
Mo method
By developing Avrami's model, which assumed that the non-isothermal crystallization process was a small step in isothermal crystallization, Ozawa's model was used to describe the non-isothermal crystallization. The fractional crystallinity (Xt) for time t is shown in the following Eq. (6).
The linear form of this equation was more commonly used as following Eq. (7).
where m and Ko were obtained from the slope and interception on the y-axis of the graph from the relationship between \(\mathit{ln}\left[-\mathit{ln}\left(1-{X}_{t}\right)\right]\) and \(\mathit{ln}\left(\varphi \right)\), respectively.
Mo method combined Avrami and Ozawa equations to describe non-isothermal crystallization process. Mo equation was as follows [22]:
where F(T) was the Mo modified crystallization rate parameter and γ was the ratio of n in the Avrami equation to m in the Ozawa equation, which was related to the crystallization dimension. A series of straight lines were plotted for a given value of relative crystallinity, as shown in Fig. 6. Then, \(F\left(T\right)\) and γ were obtained from the intercepts and slopes of the straight lines.The parameters obtained from the \(ln\varphi\) versus \(lnt\) relationship plots were presented in Table 6 for all samples.

Plots of \(lg\varphi\) versus \(lgt\) for the cold crystallization of (a) Free-cc, (b) 5NCC, (c) 11NCC, (d) 5MCC, (e) 11MCC, (f) 5NCC/11MCC and (g) 11NCC/5MCC
As shown in Table 6, F(T) of all the samples increased with the increase of Xt regardless of the presence of CaCO3 particles. This indicated that the faster the cooling rate, the higher the α' crystallinity achieved in unit crystallization time. Moreover, the γ value hardly changed with the increase of Xt in all the samples, indicating that the crystallizing mode did not change through crystallization. Mo method displayed that the 5MCC sample had a faster crystallization rate, which was consistent with the results in Fig. 1. This indicated that Mo method was suitable for describing the non-isothermal crystallization kinetics regardless of the particle size of CaCO3 particles.
Tensile properties analysis
Tensile testing was used to determine the mechanical properties of PBAT/PLA/CaCO3 composites, and the results were shown in Fig. 7a to d. As shown, the mechanical properties of the composites were increased after addition of the CaCO3 filler regardless of nano- and micro-CaCO3. A connection between the filler and the matrix ensured that external forces were transferred to the dispersed filler, so that the mechanical properties were significantly improved. The 5NCC/11MCC sample had the highest tensile and yield strengths, which probably exhibited the synergistic enhancement of nano- and micro-CaCO3 particles on PBAT/PLA blend. But the 11NCC/5MCC sample had the lowest, which was very different from the 5NCC/11MCC sample, indicating that the effective synergy was dependent on the weight ratio of nano- and micro-CaCO3 fillers.

Results of tensile and impact testing of PBAT/PLA/CaCO3 composite
Micromorphology of fracture surfaces
The SEM method was used to analyse the morphology of fracture surfaces of the samples further. The fracture surfaces from the samples in tensile testing were shown in Fig. 8.

SEM of fracture surfaces of the samples in tensile testing. (a) Free-CC, (b) 5NCC, (c) 11NCC, (d) 5MCC, (e) 11MCC, (f) 5NCC/11MCC and (g) 11NCC/5MCC
As shown in Fig. 8(a), the fracture surface of the Free-CC sample displayed the spherical PLA phase distributing in the PBAT continuous phase. A closer look revealed that the phase interfaces of PLA and PBAT were obviously separated on the cross section in Fig. 8(a), which indicated that PLA and PBAT had poor compatibility in the sample.
For the 5NCC sample, the particle size of the dispersed spherical PLA phase became smaller and deformed as show in Fig. 8(b), which indicated that the presence of nano-CaCO3 particles enhanced the compatibility between PLA and PBAT in the 5NCC sample. The 11NCC sample showed similar fracture characteristics to the 5NCC sample. In addition to less PLA phase with a large particle size being distinguished, more nano-CaCO3 particles showed aggregation as show in Fig. 8(c). Moreover, the stronger cross-linking enhanced the compatibility between PBAT and PLA, resulting in PLA grains forming when PBAT underwent large deformation as shown in Fig. 8(b) and (c).
The 5MCC and 11MCC samples had similar fracture surface as show in Fig. 8(d) and (e). More grains of PLA phase as that on the fracture surface of the Free-CC sample were observed on the fracture surfaces of the two samples. This showed that the continuous PBAT phase did not deform PLA phase when large deformation occurred, indicating that PBAT and PLA had poor compatibility at the interface and no strong interaction between them in the presence of micro-CaCO3. In contrast, the distribution of PLA phase in the 5MCC sample was more dense and the average particle size was smaller than that in the 11MCC sample. Compared with nano-CaCO3 particles, micro-CaCO3 particles had weaker cross-linking effects, which was not enough to deform the spherical PLA particles.
The fracture surfaces of the 5NCC/11MCC and 11NCC/5MCC samples containing both nano- and micro-CaCO3 particles displayed the synergetic characteristic as show in Fig. 8(f) and (g). The particle size of PLA phase was large and its shape became shuttle.
Characterization of PBAT crystallization
Figure 9 illustrated wide angle X-ray diffraction (WAXD) patterns of the PBAT/PLA/CaCO3 composites. The diffraction peaks attributed to the PLA matrix were indistinct in the Free-CC sample, which indicated that the crystallinity of the PLA phase in the sample was very low due to the influence of the PBAT phase. On the contrary, the diffraction peaks of PBAT were ob vious.The low diffraction peaks at 2θ = 17.7° and 23.1° corresponded to (010) and (100) planes of PBAT, respectively, although the crystallinity was not high. Without considering the diffraction peaks of the introduced calcium carbonate, no obvious differences related to the diffraction peaks, and peak positions were observed in the different samples. Therefore, the crystal structure of PBAT and PLA was not changed in the composites studied whether there was nano-CaCO3 particles or macro-CaCO3 particles.

WAXD patterns of PBAT/PLA/CaCO3 composites
Conclusion
Nanometer calcium carbonate of 5phr and 11phr significantly increased the melting temperature of PLA and PBAT in PLA/PBAT/CaCO3 composite materials. If used in combination with micron calcium carbonate of 5phr and 11phr, the melting temperature of PBAT further increased. However, the crystallinity of both PLA and PBAT decreased after the introduction of calcium carbonate.
Pure PLA did not crystallize at a cooling rate of -20 °C/min, while PBAT crystallized, indicating that the crystallization rate of PLA was much lower than that of PBAT under experimental conditions. PLA crystallized after mixing PLA and PBAT (such as in Free-CC), indicating that PBAT improved the crystallization rate of PLA. After further introduction of calcium carbonate, the crystallization rate of PLA decreased, and PLA in PLA/PBAT/CaCO3 only crystallized at a particularly low cooling rate (-4 °C/min). PBAT crystallized regardless of the change in cooling rate, and calcium carbonate reduced the crystallization rate of PBAT. The study on the non-isothermal crystallization behavior of the composites showed that the crystallization mode of PBAT remained unchanged, and nano calcium carbonate significantly reduced the crystallization rate of PBAT compared to micro calcium carbonate.
Both nano and micro calcium carbonate significantly improved the mechanical properties of the composites. Nano and micro calcium carbonate had a certain synergistic effect in improving the mechanical properties, but the synergistic effect depended on the ratio of the two. Observation of the fracture surface indicated that micro calcium carbonate particles had weaker cross-linking effects compared with nano calcium carbonate particles, which was not enough to deform the spherical PLA particles.
The ratio of nano-CaCO3 and micro-CaCO3 was controlled to achieve the synergistic enhancement effect between them for PBAT/PLA/CaCO3 composites, which was favorable to further reduce the cost of biodegradable plastic bags and to increase their market share. It was more effective to use nano-CaCO3 and micro-CaCO3 together to improve the melting temperature of PBAT and PLA in PBAT/PLA/CaCO3 composites than to use them alone.
References
Gill YQ, Mehdi S, Mehmood U (2022) Mater Lett 306:130881
Sun DX, Gu T, Qi XD et al (2021) Chem Eng J 424:130558
Shen S (2022) Mater Res Express 9:025308
Saeidlou S, Huneault MA, Li H et al (2012) Prog Polym Sci 37(12):1657–1677
Mulla MZ, Rahman M, Marcos B et al (2021) Molecules 26(7):1967
Jia S, Zhao L, Wang X et al (2022) Int J Biol Macromol 201(15):662–675
Razavi M, Wang SQ (2019) Macromolecules 52(14):5429–5441
Deng Y, Yu C, Wongwiwattana P et al (2018) J Polym Environ 26:3802–3816
Li K, Peng J, Turng L-S et al (2011) Adv Polym Technol 30(2):150–157
Li G, Xia Y, Mu G et al (2022) J Macromol Sci B 61(3):413–424
Jalali A, Huneault MA, Elkoun S (2016) J Mater Sci 51(16):7768–7779
Shi N, Dou Q (2015) J Therm Anal Calorim 119(1):635–642
Clizia A, Massimiliano B (2022) Chinese J Polym Sci 40(10):1269–1286
Gui H, Zhao M, Zhang S et al (2022) Foods 11(15):2252–2252
Danaya P, Phanwipa W, Khwanchat P et al (2021) Polymers 13(23):4192–4192
Yoksan R, Dang KM, Boontanimitr A et al (2021) Int J Biol Macromol 190:141–150
Xu C, Zhang X, Jin X et al (2019) J Polym Environ 27(6):1273–1284
Jacek A, Michał N (2021) Materials 14(6):1523
Gutiérrez AA, Arrieta MP, López-González M et al (2020) Materials 13(21):4910
Alakrach AM, Al-Rashdi AA, Al-Omar MK et al (2021) Materials Science Forum 1021:280–289
Sanusi OM, Benelfellah A, Papadopoulos L et al (2021) J Mater Sci 56:16887–16901
Deetuam C, Samthong C, Choksriwichit S et al (2020) Iran Polym J 19(2):103–116
Rocha DB, de Carvalho JS, de Oliveira SA et al (2018) J Appl Polym Sci 135(35):46660
Lee JM, Hong JS, Ahn KH (2019) Polym Composite 40(10):4023–4032
Han LJ, Han CY, Bian JJ et al (2012) Polym Eng Sci 52(7):1474–1484
Nekhamanurak B, Patanathabutr P, Hongsriphan N (2014) Energy Procedia 56:118–128
Martin O, Avérous L (2001) Polymer 42:6209–19
Zhang J, Tashiro K, Tsuji H et al (2008) Macromolecules 41(4):1352–1357
Kawai T, Rahman N, Matsuba G (2007) Macromolecules 40(26):9463–9469
Kalish JP, Aou K, Yang X et al (2011) Polymer 52(3):814–821
Chen X, Kalish J, Hsu SL (2011) J Polym Sci Part B Polym Phys 49(20):1446–1454
Chen L, Dou Q (2020) J Therm Anal Calorim 139(2):1069–1090
Funding
This study was supported by the Scientific Project of Luohe Medical College (Grant No. 2020-LYZZHXM010 and 2019-LYZZHYB024).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xie, J. Effect of mixing strategy on thermal and mechanical properties of poly(butylene adipate-co-terephthalate)/poly(lactic acid) incorporated with CaCO3 fillers. J Polym Res 30, 229 (2023). https://doi.org/10.1007/s10965-023-03618-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-023-03618-5