
Abstract
Homatropine Methylbromide (HMB) has been used as a template to synthesize two novel molecularly imprinted polymers (MIPs), HMB-MIP1 and HMB-MIP2, which correspond to the use of two different functional monomers (FMs), methyl acrylate (MA) and acrylamide (AM), respectively. The IR, SEM and TGA techniques are used to characterize these MIPs. Then, their binding characteristics to HMB were evaluated in terms of adsorption isotherm. Density Functional Theory (DFT) calculations were used to study the structures of each of the pre-polymerized complexes, (HMB-Complex1 and HMB-Complex2 refers for HMB-MIP1 and HMB-MIP2, respectively. The study includes calculating the complexation energies and Gibbs free energies of the two complexes. The presence and strength of the non-covalent interactions within pre-polymerized complexes were examined using Quantum Theory of Atoms in Molecules (QTAIM).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
HMB is a homatropine quaternary derivative (Fig. 1). It has an antimuscarinic action, so it is combined with a few medicines to treat gastrointestinal cramps [1]. There are several analytical methods for determining the HMB, including: turbidimetric [2], spectrophotometric [3], titration [4], and chromatographic [5].

Chemical structure of HMB
Molecularly imprinted polymers (MIPs) are porous macromolecular materials produced with a tailored binding affinity for a target chemical (Template). They act as plastic antibodies, receptors or enzymes as they have a high affinity that is comparable to their biological counterparts with the added benefit of being more durable than them [6]. The targeted template, a functional monomer (FM), a crosslinker, a polymerization initiator, and a porogen constitute the MIP components. The spatial arrangement of FMs around the template is determined by intermolecular interactions between them, the subsequent polymerization of the FMs in an excess amount of the crosslinker which acts as a glue fixes the the three-dimensional structure. After the template is removed, chemically and sterically complementary void volume like template structure is left in the polymer network that serve as sites of molecular recognition for same template [7, 8]. They have been used in promising analytical methods due to their high specificity towards their target molecules, as sorbents in matrix pretreatment, as packing material in separation stationary phases in chromatographic techniques [9,10,11,12]. The initial stage of MIP preparation involves the formation of a complex between a template and FM through non-covalent interactions in the self-assembly process and is called a pre-polymerized complex. The stability of this complex is important in this step and relies on the strong binding of the FM with the template and ultimately gives the efficient performance of the MIP properties of high binding capacity, selectivity, and affinity to its template [13]. Therefore, it is necessary to know and study the number and strength of these interactions. Computational chemistry approaches are fast, robust and rational predictive tools used to screen the possible non-covalent interactions that occur in the pre-polymerization complex at the molecular level. Thus, it enables us to define the optimum conditions to prepare the MIP with high efficiency to recognize its analyte, instead of the usual experimental method for MIP synthesis which is costly, time-consuming, laborious and choosing the ideal imprinting conditions is typically done empirically based on chemical intuition and trial-and-error methods, spending large amounts of precursors reactants in the process [14]. Density functional theory (DFT) is one of the most accurate and precision of quantum mechanics methods, is used to gain a better understanding for electronic properties of a molecule, its strategies applied on MIP included such selecting the best template/FM molar ratio and the best type of FM based on calculated binding energies of pre-polymerized complex as the difference between complex total electronic energy and the reactants energies, as well as the suitable porogen which polymerization occur in it, and offers a distinctive comprehension of thermodynamic and spectroscopic aspects for a system from a molecular level perspective [15, 16]. Herein, we report the preparation of two MIPs using HMB as a template. the two FMs (MA and AM) were used to prepare, HMB-MIP1 and HMB-MIP2. Ethylene glycol dimethacrylate (EGDMA) as a crosslinker, benzoyl peroxide (BP) as the radical initiator of polymerization, and Methanol/Chloroform (50:50, V/V) as the porogen for synthesis. After that, they were tested for rebinding to HMB. Since the pre-polymerized complex may serve as a key for predicting MIP performance. Quantum Mechanical calculations approaches as DFT is used to focus on the intermolecular interactions for the structures of the HMB-FM complex at each molar ratio. Also, the complexation energies of their pre-polymerized complexes were studied in vacuum and different solvents. Thermodynamics parameters of the production of the pre-polymorized complexes in self-assembley reaction were estimated to evaluate the spontaneity of their formation. Also, the nature of the intermolecular interactions were inspected using QTAIM.
Experimental part
Chemicals
The solvents (chloroform/methanol) which were used in the preparation of MIPs are obtained from Merck (Darmstadt, Germany), while the other chemicals were purchased from Sigma-Aldrich (St. Louis-USA). The state corporation of drug and medical appliances (SDI-Samarra, Iraq) provided a reference of HMB as a grant.
Instrumentation
The prepared MIPs are examined using scanning electron microscopy (SEM) (TESCAN MIRA3-Czech Republic). UV–Visible Spectrophotometer 1800 PC (SHIMADZU-Japan) was used to perform measurements relevant to HMB determination in their solutions employing (1.0 cm) quartz cells. IRAffinity-1S (SHIMADZU-Japan) was used to record the infrared spectra. The polymer mixture is heated and agitated using an ultrasonic sensitive water bath from (SONOREX-GERMANY). Thermogravimetric (TGA) analysis were carried out between 25 and 600 °C using a maximum heating rate of 20 °C / min. in a nitrogen environment, using the SDT Q600 V20.9 Build 20 (TA Instruments, USA).
MIP synthesis
In two glass vials, two HMB-MIPs (HMB-MIP1 and HMB-MIP2) were synthesized using the two FMs MA and AM. The following chemicals were added into each vial: (0.27 mmol) of HMB, (1.95) mmol MA in first vial and (2.8 mmol) AM in the second vial as the FM. Then, (19.67 mmol) of EGDMA were added to 4.0 mL of methanol, and to this mixture (1.24 mmol) of BP was dissolved in 4.0 mL of chloroform was added into each vial to uniformly homogenize this mixture, the solutions were sonicated while they where purged with N2 gas in an ice/water slurry for 5 min to remove oxygen and, then, sealed tightly, after that they were moved into a different water bath at a higher temperature (60 °C) until polymerization completed. The polymerization time of HMB-MIP1 took longer than HMB-MIP2, about 24 h for the first, while about 15 h for the latter. The two synthesized polymers were rinsed using CH3OH and dried, then crushed using a mortar and pestle. The crushed samples were eluted from HMB in a Soxhlet using the following solvent mixture (methanol;acetic acid;distilled water:70;20;10) for several times until HMB disappeared entirely. The absorbance values were measured at 205 nm which indicated that HMB has been eluted. the final product are left at RT for slow drying.
Isothermal rebinding tests
0.2 and 0.4 g of each HMB-MIP mixed with 10 mL HMB solution of concentrations ranged from 10 to 40 ppm were incubated at 25 °C to assess their ability to bind to HMB. After a spectrophotometric measurement of the unbound HMB concentration in the supernatant at 205 nm, the binding capacity Q (μMole.g−1) was calculated using the following equation:
where V is the portion volume (mL), Cinit is the initial concentration (mMole.L−1), Cfree is the free final concentration of HMB and mMIP is the mass of the polymer particles (mg). Values from the isothermal equilibrium are used to decide wheather the Freundlich (Eq. 2) or Langmuir (Eq. 3) isotherm model by depending on the correlation coefficient (R2).
where Ce is the equilibrium concentration of HMB in solution, qe is the quantity of HMB adsorbed per mass unit of adsorbent, kf depicting the adsorption capacity, and n is the adsorption intensity, qm denotes the maximum adsorption quantity of monolayer model, kL denotes the Langmuir constant.
Subsequently, the results of the experiment on static adsorption were processed using Scatchard's Analysis (Eq. 4) to screen the HMB-MIPs binding parameters:
The maximum adsorption capacity is represented by Qmax, whereas Kd is the dissociation constant at the binding site.
Theoretical part
Geometrical optimization of molecules
Molview.org was used to get the structures of the compounds HMB, MA and AM. All of our computational studies were carried out at DFT/B3LYP level and 6-31G as basis set. Using G09 [17], the structures of pre-polymerized complexes have optimized (S1). The optimized structures are ground true minima since there are no imaginary frequency for each of them. Addition, The molecular electrostatic potential surface (MEP) [18] has been used as a tool for mapping electron-rich and electron-poor sites to facilitate understanding of how the intermolecular interactions of complex formation occur between HMB-FMs.
Computational screening of the formed complexes between HMB and FMs
A computational study at the same theoretical level mentioned above was conducted to screen and establish stable configurations of different molar ratios for complexes arising between HMB and FM, which were based on strong overlaps between the electron-rich and electron-poor sites between them. Multiple interactions resulting in each configuration were also taken into account. We calculated the complexation energy for each of the ratios using the mathematical relationship as following:
where FMs represents the number of MA or AM that contributed to the molar ratio. Besides this, thermodynamic parameters [19] the following eqs.:
where E is the total electronic energy, the Gibbs free energy correction is Gcorr and the enthalpy correction is Hcorr.
Simulation in reaction media
Whereas the complexation energy was calculated in vacuum, solvents are known to reduce the interaction within FM-Template complex. Hence, it would be preferable if the solvent as reaction medium would possess the strongest intermolecular forces. When selecting a solvent as a porogen in MIP preparation, there are several aspects have to be considered. In terms of preparation, all components must be soluble and not to hinder the interactions between the FMs and template in the pre-polymerized complex FM-Template, but in terms of rebinding, the polarity of porogen in the imprinting process have to be preferably similar to the polarity of the aqueous matrix for analyte in order to maintain the microenvironment consistency that it can make it simple to rebind [20, 21].
QTAIM topology analysis
The QTAIM is one of the most popular and validated techniques for non-covalent interaction analysis [22, 23], which interprets weak interaction features based on the characteristics of the interatomic bond critical points (BCPs) [24]. These points are saddle points along the bond path, which the electronic density at it begins to increase gradually until it terminates at the nucleus, so these points are considered the most depictive points for interatomic interaction atoms [25, 26]. AIMALL program was used to carry out the QTAIM analysis [27].
Results and discussion
Characterization of HMB-MIPs: FT-IR spectra, morphology & TGA
The comparative FT-IR spectra of the HMB and HMB-MIPs are shown in Figs. 2 and 3. The values for the distinct peaks and their changes are listed in Tables 1 and 2. The HMB-MIPs spectra revealed three significant HMB peaks whose variations could be seen. First, for the stretching OH group. Next, for the stretching of (C-H) and (C = C) groups associated with the aromatic system of HMB, whose changes can be seen. The leached HMB-MIPs lack all of the foregoing HMB peaks, which indicates that the prepared MIPs were successful in eluting HMB from them. SEM has been used to analyze polymer morphology. The results show that the surfaces of the washed HMB-MIPs (Figs. 4b and 5b) were rough and less uniform, unlike the surfaces of the unwashed HMB-MIPs (Figs. 4a and 5a). Thus, the surfaces of the washed HMB-MIPs are capable of adsorbing HMB from its solutions.

FT-IR spectra of HMB, Unleached HMB-MIP1 & leached HMB-MIP1

FT-IR spectra of HMB, Unleached HMB-MIP2 & leached HMB-MIP2

SEM of HMB-MIP1

SEM of HMB-MIP2
For TGA measurements of samples (S2), once heating started, all curves have a significant moisture loss. For a HMB, at 225 °C it begin to decompose up to 350 °C.
The TGA thermograms of unleached and leached HMB-MIP1. The unleached started to decompose at lower temperatures which indicates that leached form is more thermaly stable. In the unleached, first the interactions between the HMB and MA monomers are broken at the binding cavities and the liberated molecules of monomer and crosslinker in the polymer begin to decompose [28]. At 240 °C the decomposition of the HMB begins, and about 270 °C the backbone of polymers decompose up to 425 °C, finally the process decomposition of polymers were completed from 475 to 525 °C. Unlike in leached HMB-MIP1, it differs from it only in that the decomposition of the polymer here took place in one stage, due to the absence the HMB in it. While in an unleached HMB-MIP2, at 225 °C the interactions between the HMB and AM monomers are broken and molecules of crosslinker and monomers are released, as well as ammonia and water vapors were evolved, by the imidization process [29] and at 230 °C the decomposition of the HMB begins. After that, about 300 °C up to 450 °C is attributed to the breakdown of backbone polymers and imides formed in the previous step. In contrast to in leached HMB-MIP2, it differs from it in the presence of two decomposition regions of the polymer, where the absence of HMB causes an increase in imidization reactions, which results in a higher heat resistance and a loss of initial weight of the polymer less than in the unleached. Eventually, we conclude that the HMB-MIP2 gives more stable thermal properties than the HMB-MIP1.
Active sites for individuals of pre-polymerized complexes
Each of the molecules that contributed to the construction of the HMB-FM complex, namely the HMB, MA, and AM, had their optimal conformers determined, and after that, using MEP, the active sites for each of these molecules were examined. These structures are presented in Figs. 6, 7 and 8.

Structure & MEP of HMB

Structure and MEP of MA

Structure & MEP of AM
The active sites (electron rich sites, red color in Figs. 6, 7 and 8) in the HMB are (Br1, O2, O19 and, O17) act as proton acceptors, and (the electron deficient sites, blue color in Figs. 6, 7 and 8) (H18, H45, H46, H26, H37, H41, H23, H32, H21, H36, H5, H8 and H28) act as proton donors. While in MA FM, the proton acceptors are (O1 and O2) and (H7, H8 and H9) are the proton donors. Finally, in an AM monomer, the proton acceptor is (O7) and (H9 and H10) are the proton donors.
Computational study for molar ratio of HMB-complexes
Under same theoretical level B3LYP/6-31G, various molar ratios were established to form stable complexes. All ratios are depicted in Figs. 9 and 10. We started with a ratio of 1:1, with gradually increasing the complexation energy continuing with it until the increase in energy after that was trivial due to the intermolecular interaction of the FM with itself reducing the selectivity [30]. As a result, this can be considered as a theoretical analysis [31] to determine the exact stoichiometric molar ratio between template and FM. Details of all the generated complexes are given in Tables 3 and 4. The complexes that formed between the HMB and the MA were obtained by two types of interactions. First, between the oxygen of carbonyl for MA interact with the hydrogen atoms in the HMB. Second, those intercations formed between the hydrogen of methyl for MA and the counter bromide ion of HMB or the oxygen atoms in the structure of the HMB. Whereas for the interactions that occur between the HMB and the AM, there are also two types; first, between the oxygen of carbonyl for AM and the hydrogen atoms in the HMB and second, those that were between the hydrogen of amine and the counter bromide ion or the oxygen atoms in the structure of the HMB. Comparing the results that are listed in Tables 3 and 4, we find that the binding energies of the complexes resulting from AM and the HMB are much stronger than in MA with the HMB, this is due to the strength of charge distribution in AM compared to MA, as we have seen in the MEP of the molecules in Figs. 7 and 8.

Configurations for Molar Ratios of HMB to Mas

Configurations for Molar Ratios of HMB to AMs
The resultant stoichiometric molar ratios of 1 to 7 and 1 to 10 correspond to HMB-Complex1 and HMB-Complex2. The latter had a higher molar ratio because the HMB required more AM to generate the stable complex in the end. The higher complexation energy of the HMB-Complex2 is also attributed to the strength of the interactions as well as to the abundance of multiple hydrogen bonding, not merely due to its greater molar ratio. Moreover, from thermodynamic parameters shown in Tables 5 and 6, the formation process of pre-polymerized HMB-Complexes is exothermic, since the enthalpy values (ΔH°) are negative. The enthalpy values are -57.58, -152.97 kcal.mol−1 for HMB-Complex1, and HMB-Complex2, respectively.
The negative entropy, ΔS°, values associated the formation of the pre-polymerized complexes which are -251.94, -363.05 cal.K−1.mol−1 for HMB-Complex1 and HMB-Complex2, respectively, indicates that the non-covalent interactions that have occurred throughout the imprinting process result in a transformation to more ordered state. The predicted ΔG° value for the HMB-Complex1 is positive + 17.53 kcal.mol−1, suggesting that the pre-polymerization complex is not thermodynamically favored (equilibrium constant, K < 1). As a result, stirring [32] is necessary to achieve the imprinting process. Also, MIPs are typically processed at mild temperatures between 50 and 60 °C, as heating above these causes the complex to lose its structural integrity. While a negative value for HMB-complex2 of -44.73 kcal.mol−1 indicates that AM facilitates the preparation of pre-polymerization complexes from a thermodynamic aspect. These results indicate that AM has greater affinity than MA, so it has the opportunity to be used as FM to synthesize MIP having better recognition properties on HMB.
Evaluating of selected porogen
Based on the simulation results for both pre-polymerized complexes, HMB-Complex1 and HMB-Complex2 in solvents as shown in Tables 7 and 8, it was found that the non-polar solvent has the highest complexation energies, while among the polar solvents, methanol had the higher complexation energy, more than both acetonitrile and DMSO, because it was revealed that increasing the value of the dielectric constant ε of the solvent with it the value of complexation energy decreases [14]. Therefore, the porogen used as a mixture between two immiscible solvents that differ in polarity was a good choice in the preparation of the HMB-MIPs in terms of experimental and theoretical. First, experimentally methanol dissolves HMB and improves the rebinding process because it provides a solvation environment for the imprinting process similar to that of HMB's solutions [33], and theoretically has a higher complexation energy value among polar solvents. The second is chloroform, a nonpolar solvent that has a weak effect on complexes between HMB and FMs, so theoretically it has the highest complexation energy value than any others.
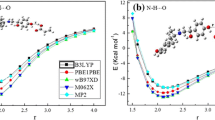
QTAIM's interpretation of intermolecular interactions
The QTAIM study provides the following values: total electronic energy density H(r), Laplacian of electronic density ∇2ρ(r), potential energy density V(r), and kinetic energy density G(r). With the help of these parameters, the types of chemical interaction and their extents can be described. Also, the energy value of each interaction was estimated using Eint = V(r)/2 [34]. All values of ∇2ρ(r) were positive based on the results reported in Tables 9 and 10, which means that these interactions are closed-shell interactions or non-covalent interactions [35]. The nature of these interactions could be defined in terms of the values of the following parameters ρ(r), ∇2ρ(r) and Eint. In HMB-Complex1, the value are the highest at BCP H18 – O108 which are 0.03337 a.u., 0.121604 a.u., and -9.453 kcal.mol−1 for values of ρ(r), ∇2ρ(r), and Eint, respectively. This indicates that the interaction is stronger than others, this is supported by shorter interaction distances in comparison to the other interactions. The lowest values of ρ(r), ∇2ρ(r) and Eint occur at BCP C7–H114, the values are 0.003058 a.u., 0.009689 a.u., and -0.395 kcal.mol−1 for values of ρ(r), ∇2ρ(r), and Eint, respectively, implying that the interaction was weaker than others which is attributed to its longer interaction distances. The interactions are stronger in are in HMB-complex2, the highest values of ρ(r), ∇2ρ(r), and Eint occur at BCP H18-O123 which are 0.052787 au, 0.177764 au, and -15.1 kcal.mol−1, respectively, and the lowest values of ρ(r), ∇2ρ(r), and Eint occur at BCP H5-O103 which are 0.003393 a.u., 0.013952 a.u., and -0.559 kcal.mol−1, respectively.
Adsorption isotherms and scatchard analysis
The plots shown in (Figs. 11 and 12) and the data listed Tables 11 and 12 indicate that as Ci of HMB increased, Q increased along with it. Based on the Table 13 for the HMB-MIPs' experimental results, the Langmuir fitting model is proved to be more appropriate. This demonstrated that the hydrogen interactions between HMB and the specific binding sites on the surfaces of the HMB-MIPs were responsible for the monolayer adsorption. Also, we found that Q values in HMB-MIP2 are greater than HMB-MIP1. which indicates that the pre-polymerized complex HMB-Complex2 is more robust. For the scatchard analysis (Fig. 13), it revealed linear and a single straight line at both HMB-MIPs at 0.4 g, indicating homogeneity and affinity of their binding sites. The Kd and Qmax values for HMB-MIP1 were 0.0094 mM and 2.64 μmole.g−1, respectively. Whereas, with HMB-MIP2, Kd and Qmax were 0.012 mM and 3.2 μmole.g−1, respectively.

Binding Isotherm for HMB-MIPs at 0.2 g

Binding Isotherm for HMB-MIPs at 0.4 g

Scatchard Analysis of HMB-MIPs at 0.4 g
Conclusion
In conclusion, depending on the bulk polymerization, two types FMs, MA and AM were used to synthesize MIPs namely, HMB-MIP1 and HMB-MIP2 for HMB recognition. Understanding the mechanism of the imprinting process, describing how molecular interactions are constructed, study the potential of solvents used in the MIPs preparation and accessing their pre-polymerization complexes: HMB-Complex1 and HMB-Complex2 as the descriptor the performance MIPs, all these important points were crucially achieved through computational chemistry representing by DFT method. According to the theoretical results, the values for complexation energy and Gibbs free energy were -171.6, -44.73 kcal.mol−1 for HMB-Complex2 that were more preferable than -69.27, + 17.53 kcal.mol−1 of HMB-Complex1. Also, the data obtained by QTAIM indicates to strength of the interactions of HMB-Complex2. In terms of experiments, there were very compatible with theoretical results, compared to HMB-MIP1, we found that HMB-MIP2 had higher binding capacity Q values. As well as, TGA mesurements were proved that HMB-MIP2 was most thermally stable than HMB-MIP1 due to the strong interactions resulting from the use AM as monomer in its preparation. Therefore, HMB-MIP2 had stronger recognition properties on HMB than HMB-MIP1 to adsorb HMB from its aqueous solutions.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
Ebadi M (2007) Pharmacodynamic Basis of Herbal Medicine, 2nd edn. CRC Press, New York, p 207
Canaes LS, Leite OD, Fatibello-Filho O (2006) Flow-injection turbidimetric determination of homatropine methylbromide in pharmaceutical formulations using silicotungstic acid as precipitant reagent. Talanta 69:239–242. https://doi.org/10.1016/j.talanta.2005.09.035
Hanna S, Rosen M, Rasero L, Lachman L (1977) Analysis of homatropine methylbromide dosage forms. J Pharm Sci 66:123–124. https://doi.org/10.1002/jps.2600660135
The United States Pharmacopeia (1995) The National Formulary, USP-23, NF-18, p. 744
Majlat P, Helboe P, Kristensen AK (1981) Separation and quantitative determination of homatropine methylbromide and opium alkaloids in admixture in pharmaceutical preparations by gas-liquid and high-performance liquid chromatography. Int J Pharm 9:245–255. https://doi.org/10.1016/0378-5173(81)90050-8
Sullivan MV, Dennison SR, Archontis G, Reddy SM, Hayes JM (2019) Towards rational design of selective molecularly imprinted polymers (MIPs) for proteins: Computational and experimental studies of acrylamide-based polymers for myoglobin. J Phys Chem B 123(26):5432–5443. https://doi.org/10.1021/acs.jpcb.9b03091
Vasapollo G, Del Sole R, Mergola L, Lazzoi MR, Scardino A, Scorrano S, Mele G (2011) Molecularly imprinted polymers: Present and future prospective. Int J Mol Sci 12(9):5908–5945. https://doi.org/10.3390/ijms12095908
Pardeshi S, Patrikar R, Dhodapkar R, Kumar A (2012) Validation of computational approach to study monomer selectivity toward the template Gallic acid for rational molecularly imprinted polymer design. J Mol Model 18:4797–4810. https://doi.org/10.1007/s00894-012-1481-5
Zhu Fang, Li Luwei, Xing Junde (2017) Selective adsorption behavior of Cd(II) ion imprinted polymers synthesized by microwave-assisted inverse emulsion polymerization: Adsorption performance and mechanism. J Hazard Mater 321:103–110. https://doi.org/10.1039/C6CS00061D
Zhu F, Yanhong Lu, Ren T, He S, Gao Y (2017) Synthesis of ureido-functionalized Cr(VI) imprinted polymer: adsorption kinetics and thermodynamics studies, Desalin. Water Treat 100:126–134. https://doi.org/10.5004/dwt.2017.21683
Zhu F, Li L, Li N, Liu W, Liu X, He S (2021) Selective solid phase extraction and preconcentration of Cd(II) in the solution using microwave-assisted inverse emulsion-suspension Cd(II) ion imprinted polymer. Microchem J 164:106060:1–9. https://doi.org/10.1016/j.microc.2021.106060
Chen L, Wang X, Wenhui Lu, Xiaqing Wu, Li J (2016) Molecular imprinting: perspectives and applications. Chem Soc Rev 45:2137–2211. https://doi.org/10.1039/C6CS00061D
Chianella I, Karim K, Piletska EV, Preston C, Piletsky SA (2006) Computational design and synthesis of molecularly imprinted polymers with high binding capacity for pharmaceutical applications-model case: Adsorbent for abacavir. Anal Chim Acta 559:73–78. https://doi.org/10.1016/j.aca.2005.11.068
Silva CF, Borges KB, Nascimento CS Jr (2019) Computational study on acetamiprid-molecular imprinted polymer. J Mol Model 25(104):1–5. https://doi.org/10.1007/s00894-019-3990-y
Hammam MA, Abdel-Halim M, Madbouly A, Wagdy HA, El Nashar RM (2019) Computational design of molecularly imprinted polymer for solid phase extraction of moxifloxacin hydrochloride from Avalox® tablets and spiked human urine samples. Microchem J 148:51–56. https://doi.org/10.1016/j.microc.2019.04.063
Suryana S, Mutakin YR, Hasanah AN (2021) An update on molecularly imprinted polymer design through a computational approach to produce molecular recognition material with enhanced analytical performance. Molecules 26(7):1891. https://doi.org/10.3390/molecules26071891
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian 09, Revision D.01, Gaussian Inc, Wallingford, CT
Lakshminarayanan S, Jeyasingh V, Murugesan K, Selvapalam N, Dass G (2021) Molecular electrostatic potential (MEP) surface analysis of chemo sensors: An extra supporting hand for strength, selectivity & nontraditional interactions. J Photochem Photobiol 6:1–5. https://doi.org/10.1016/j.jpap.2021.100022
Xie L, Nan Xiao Lu, Li XX, Li Y (2019) An investigation of the intermolecular interactions and recognition properties of molecular imprinted polymers for deltamethrin through computational strategies. Polymers 11(11):1872. https://doi.org/10.3390/polym11111872
Azizi Ali, Bottaro Christina S (2019) A critical review of molecularly imprinted polymers for the analysis of organic pollutants in environmental water samples. J Chromatogr A 1614:460603. https://doi.org/10.1016/j.chroma.2019.460603
Sales TA, Ramalho TC (2020) Computational design of synthetic receptors for drug detection: interaction between molecularly imprinted polymers and MDMA(3,4-methylenedioxymethamphetamine). Theoret Chem Acc 139(31):1–12. https://doi.org/10.1007/s00214-020-2543-x
Ebrahimi A, Karimi P, Akher FB, Behazin R, Mostafavi N (2014) Investigation of the π–π stacking interactions without direct electrostatic effects of substituents: the aromatic∥aromatic and aromatic∥anti-aromatic complexes. Mol Phys 112:1047–1056. https://doi.org/10.1080/00268976.2013.830784
Gorokh ID, Adonin SA, Novikov AS, Usoltsev AN, Plyusnin PE, Korolkov IV, Sokolov MN, Fedin VP (2019) Halobismuthates with 3-iodopyridinium cations: halogen bonding-assisted crystal packing. Polyhedron 166:137–140. https://doi.org/10.1016/j.poly.2019.03.041
Lane JR, Contreras-García J, Piquemal J-P, Miller BJ, Kjaergaard HG (2013) Are bond critical points really critical for hydrogen bonding? J Chem Theory Comput 9(8):3263–3266. https://doi.org/10.1021/ct400420r
Matta CF, Arabi AA (2011) Electron-density descriptors as predictors in quantitative structure–activity/property relationships and drug design. Future Med Chem 3(8):969–994. https://doi.org/10.4155/fmc.11.65
Pilmé J, Renault E, Bassal F, Amaouch M, Montavon G, Galland N (2014) QTAIM analysis in the context of quasirelativistic quantum calculations. J Chem Theory Comput 10(11):4830–4841. https://doi.org/10.1021/ct500762n
Todd A, Keith TK (2015) AIMAll (Version 15.05.18), Gristmill Software, Overland Park KS. aim.tkgristmill.com
Farzaneh S, Asadi E, Abdouss M, Barghi-Lish A, Azodi-Deilami S, Khonakdar HA, Gharghabi M (2015) Molecularly imprinted polymer nanoparticles for olanzapine recognition: application for solid phase extraction and sustained release. RSC Adv 5(12):9154–9166. https://doi.org/10.1039/C4RA12725K
Leung WM, Axelson DE, Van Dyke JD (1987) Thermal degradation of polyacrylamide and poly(acrylamide-co-acrylate. J Polym Sci Part A-1 Polym Chem 25(7):1825–1846. https://doi.org/10.1002/pola.1987.080250711
Zhang B, Fan X, Zhao D (2019) Computer-aided design of molecularly imprinted polymers for simultaneous detection of clenbuterol and its metabolites. Polymers 11(1):1–21. https://doi.org/10.3390/polym11010017
Khan MS, Pal S (2018) Quantum mechanical studies on dioxin-imprinted polymer precursor composites: Fundamental insights to enhance the binding strength and selectivity of biomarkers. J Mol Recognit 31(11):e2736. https://doi.org/10.1002/jmr.2736
Zhang P, Ji X, Zhang H, Xia B (2017) Quantum investigation into intermolecular interactions between bisphenol A and 2-Vinyl/4-vinylpyridine: Theoretical Insight into molecular imprinting complexes. Comput Theor Chem 1108:76–85. https://doi.org/10.1016/j.comptc.2017.03.025
Spivak DA (2005) Optimization, evaluation, and characterization of molecularly imprinted polymers. Adv Drug Deliv Rev 57(12):1779–1794. https://doi.org/10.1016/j.addr.2005.07.012
Espinosa E, Molins E, Lecomte C (1998) Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem Phys Lett 285:170–173. https://doi.org/10.1016/S0009-2614(98)00036-0
Kazemi S, Daryani AS, Abdouss M, Shariatinia Z (2016) DFT computations on the hydrogen bonding interactions between methacrylic acid-trimethylolpropane trimethacrylate copolymers and letrozole as drug delivery systems. J Theor Comput Chem 15:1650015. https://doi.org/10.1142/S0219633616500152
Acknowledgements
The authors express their gratitude and appreciation to the Department of Chemistry, College of Science, Baghdad University, Iraq, for using their equipment and facilities.
Author information
Authors and Affiliations
Contributions
Experimental design: Yehya K.Al-Bayati; Experimental work: Mustafa J. Bichan; Theoretical accounts and Molecular Modeling: Firas F.Awwadi and Mustafa J. Bichan; data analysis: Faiz M.AL-Abady and Mustafa J. Bichan; writing-original draft preparation: Firas F.Awwadi and Mustafa J. Bichan; writing-review and editing: Faiz M.AL-Abady and Mustafa J. Bichan.All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
On behalf of all authors, Mustafa J. Bichan states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bichan, M.J., Al-Bayati, Y.K., AL-Abady, F.M. et al. Theoretical study of molecularly imprinted polymers prepared for homatropine methylbromide. J Polym Res 30, 142 (2023). https://doi.org/10.1007/s10965-023-03513-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-023-03513-z