
Abstract
Stereocomplex polylactides (scPLAs) are high-performance biodegradable bioplastics because they show better mechanical properties and heat-resistance than the poly(L-lactide) (PLLA). However, scPLAs retain low flexibility similar to that of PLLA. In this work, a flexible poly(L-lactide)-b-poly(ethylene glycol)-b-poly(L-lactide) (PLLA-PEG-PLLA/PDLA) was melt blended with poly(D-lactide) (PDLA) before compression molding to form scPLA films. The effect of chain extension was investigated. Stereocomplexation of PLLA-PEG-PLLA/PDLA blends improved with PDLA ratio. The thermal stability of both the blends with and without chain extension was improved by stereocomplex formation. The non-chain-extended blend films became very brittle when 40 wt% PDLA was blended. Chain extension enhanced the tensile properties of the blend films. Heat resistance of blend films was significantly improved when the PDLA ratio was increased up to 20 wt%. Dimensional stability to heat at 80 °C of the film samples confirmed the results of heat resistance. The melt-processed scPLA in this work may offer revolutionary improvements in flexible and high heat-resistant bioplastics.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Poly(L-lactide) or poly(L-lactic acid) (PLLA) is an important bioplastic due to its bio-renewability, non-toxicity, biocompatibility, biodegradability and good processing properties [1,2,3]. PLLA has been widely investigated for use in many applications, such as tissue engineering, drug delivery and packaging [4,5,6]. However, its poor heat-resistance due to poor crystallizability and its brittleness due to a high glass-transition temperature (Tg ≈ 60 °C) have limited some of its applications [7, 8].
The crystallization rate of PLLA was accelerated by blending with poly(D-lactide) (PDLA) form as stereocomplex polylactides (scPLAs) that enhanced its heat-resistant properties [9,10,11]. Stronger interactions in stereocomplex crystallites of the scPLA induced higher melting temperatures (Tm ≈ 220–240 °C) and faster crystallization speed than the PLLA [12, 13]. These properties improved the heat and hydrolysis resistance of scPLA. The scPLA with high heat-resistance is appropriate for specific applications such as heat-treatment packaging, hot-fill packaging and microwave applications. However, the scPLA was still brittle because its Tg was similar to that of PLLA.
Poly(ethylene glycol) (PEG) and poly(propylene glycol) have been used as plasticizers to improve scPLA flexibility by decreasing the Tg of scPLA [14,15,16]. However, the migration of plasticizers makes scPLA unstable after aging [17]. PEGs have been used as macro-initiators for synthesizing the flexible PLLA-PEG-PLLA triblock copolymers to prevent the migration effect. Triblock copolymers of PDLA-PEG-PDLA have been synthesized for solution blending with the PLLA [18,19,20,21,22,23] and PLLA-PEG-PLLA [24] to improve flexibility of obtained scPLAs. The flexible PEG middle-blocks of both the PLLA-PEG-PLLA and PDLA-PEG-PDLA enhanced the plasticizing effect.
In a previous work showed that high molecular-weight (M.W.) PLLA-PEG-PLLA exhibited highly extensibility by chain-extension reaction [25]. They were highly flexible bioplastics. However their heat-resistance was still poor. To the best of our knowledge, the PLLA-PEG-PLLA/PDLA blends with and without chain-extension reaction have not been reported so far. Moreover, fabrication of the scPLA by melt process is very interesting at industrial scale. It is known that the stereocomplexation of PLA strongly depended on the M.W. of both the PLLA and PDLA [26, 27]. PLLA and PDLA with low M.W. had easier chain mobility for stereocomplex formation [28, 29]. Therefore in this work, the low M.W. PDLA was selected to induce stereocomplex formation of the flexible PLLA-PEG-PLLA. The influences of blend ratio and chain extension on stereocomplexation, thermal decomposition, tensile properties and heat resistance of PLLA-PEG-PLLA/PDLA blends was investigated in this research.
Experimental
Materials
L-lactide (LLA) and D-lactide (DLA) monomers were synthesized from 85 wt% L-Lactic acid (L-form >95%, Purac, Thailand) and 80 wt% D-lactic acid (D-form >99%, Haihang Industry (Jinan) Co., Ltd., China) respectively, by direct polycondensation before thermal de-polymerization. The crude LLA and DLA monomers were purified by repeated re-crystallization from ethyl acetate before drying in a vacuum oven at 50 °C for 24 h. PEG (M.W. = 20,000 g/mol, Sigma-Aldrich, Switzerland) macro-initiator was dried in a vacuum oven at 50 °C for 24 h. 1-Dodecanol (98%, Fluka, Switzerland) initiator was purified by fractional distillation under reduced pressure and stored over molecular seives at room temperature. Stannous octoate (Sn(Oct)2, Sigma-Aldrich, Switzerland) catalyst was used as supplied. Joncryl® ADR 4368 chain extender (BASF, Thailand) was used without further purification.
Synthesis and characterization of PLLA-PEG-PLLA and PDLA
Ring-opening polymerization of PLLA-PEG-PLLA and PDLA were carried out in bulk at 165 °C under a dry nitrogen atmosphere for 6 h and 2.5 h, respectively, using 0.075 mol% and 0.01 mol% Sn(Oct)2 catalyst, respectively. The PEG and 1-dodecanol were used as initiators for synthesizing the PLLA-PEG-PLLA and PDLA, respectively. PDLA with a theoretical M.W. of 5000 g/mol was synthesized using 2.8 mol% 1-dodecanol. The theoretical M.W. of PLLA-PEG-PLLA was approximately 120,000 g/mol. The obtained PLLA-PEG-PLLA and PDLA were granulated into small pieces (~5 mm) and dried in a vacuum oven at 110 °C for 3 h.
PLLA-PEG-PLLA and PDLA were characterized by gel permeation chromatography (GPC, Waters e2695 separations module) to measure number-average molecular weight (Mn) and dispersity (Ð) as well as differential scanning calorimetry (DSC, Perkin-Elmer Pyris Diamond) to observe their Tg and Tm. The characteristics of PLLA-PEG-PLLA and PDLA are summarized in Table 1.
Preparation of PLLA-PEG-PLLA/PDLA blends
PLLA-PEG-PLLA, PDLA and Joncryl® were dried in a vacuum oven at 50 °C overnight before melt blending using an internal mixer (HAAKE Polylab OS System) at 200 °C for 4 min. A rotor speed of 100 rpm was chosen. Blends with PLLA-PEG-PLLA/PDLA ratios of 100/0, 90/10, 80/20, 70/30 and 60/40 (w/w) were investigated. The Joncryl®-free and 4.0 phr Joncryl® blend series were prepared. The obtained blends were granulated into small pieces (~5 mm) and dried in a vacuum oven at 50 °C overnight before characterization and compression molding.
The blend films were fabricated using a compression molding machine (Auto CH Carver) at 240 °C for 1.0 min without any force followed with a 5.0 ton compression force for 1.0 min before quickly cooling. The films with 0.2–0.3 mm thickness were obtained and stored at room temperature for 24 h before characterization.
Characterization of PLLA-PEG-PLLA/PDLA blends
The thermal transitions of the blends were observed by means of DSC using a Perkin-Elmer Pyris Diamond DSC. For DSC heating scan, samples of 3–5 mg in weight were held at 250 °C for 2 min under a nitrogen flow to eliminate their thermal history. Then, the samples were quenched to 0 °C before heating at 10 °C/min over a temperature range of 0 to 250 °C in order to observe their Tg, Tm and cold-crystallization temperature (Tcc). For DSC cooling scan, the sample was held at 250 °C for 2 min to remove thermal history before cooling to 0 °C at a rate of 10 °C/min to observe their crystallization temperatures (Tc).
The degree of crystallinity from DSC (Xc,DSC) for homo-crystallites (hc-Xc,DSC) (Eq. 1) of the PLLA-PEG-PLLA was calculated from the enthalpy of melting for homo-crystallites (∆Hm,hc) and enthalpy of cold crystallization (∆Hcc).
where the ∆Hm,hc for 100% hc-Xc,DSC was 93 J/g [30]. The weight fraction of the PLLA (WPLLA) determined from 1H-NMR and was 0.83 for the PLLA-PEG-PLLA [25].
The Xc,DSC for stereocomplex crystallites (sc-Xc,DSC) (Eq. 2) of the PLLA-PEG-PLLA/PDLA blends was calculated from the enthalpy of melting for stereocomplex-crystallites (∆Hm,sc).
where the ΔHm,sc for 100% sc-Xc,DSC was 142 J/g [30]. The weight fractions of the PLLA and PDLA (WPLA) were 0.847, 0.864, 0.881 and 0.898 for the 90/10, 80/20, 70/30 and 60/40 PLLA-PEG-PLLA/PDLA blends, respectively.
The thermal decomposition behavior of the blends was determined by thermogravimetric analysis (TGA) using a TA-Instrument SDT Q600 TGA. For TGA, 5–10 mg of the sample was heated at 20 °C/min from 50 to 600 °C under a nitrogen flow.
The crystalline structure of the film samples was determined by wide-angle X-ray diffractometry (XRD) using a Bruker D8 Advance XRD at 25 °C operated at 40 kV and 40 mA CuKα radiation. For XRD, the film samples were recorded in a 2θ range 5° to 30° at a scan rate of 3°/min. The crystallinity from XRD (Xc,XRD) for homo-crystallites (hc-Xc,XRD) (Eq. 3) and stereocomplex crystallites (sc-Xc,XRD) (Eq. 4) of the film samples were calculated [31].
where Shc and Ssc are the integrated intensity peaks for homo-crystallites and stereocomplex crystallites, respectively. Sa is the integrated intensity of the amorphous halo.
The tensile properties of the film samples were analysed using a Lloyds LRX+ Universal Testing Machine at 25 °C. The film samples (100 × 10 mm) were determined with an initial distance between the grips of 50 mm at an extension speed of 50 mm/min according to ASTM D882. Each film sample was evaluated in at least five replicate experiments.
The thermo-mechanical properties of the film samples (5 × 20 × 0.2 mm) were measured by dynamic mechanical analysis (DMA) using a TA Instrument Q800 DMA under a tensile mode with the scan amplitude of 10 μm and the scanning frequency of 1 Hz from 40 to 140 °C at a heating rate of 2 °C/min.
The dimensional stability to heat of film samples with 0.2 mm thickness was observed by hanging the sample in an air oven at 80 °C for 30 s under a 200 g weight attached. Initial gauge length of film samples was 20 mm. The dimensional stability was calculated from the Eq. 5 [32].
Results and discussion
Thermal transitions
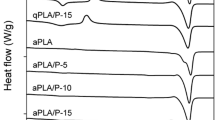
Figure 1 shows DSC heating curves of PLLA-PEG-PLLA and blends. The corresponding DSC data are reported in Table 2. The Tg of PLLA-PEG-PLLA and blends was in the range 31–33 °C and were lower than values for the PLLA [56 °C, see Fig. S1 (a)]. This is due to flexible PEG middle-blocks acting as plasticizers to decreasing Tg of PLLA end-blocks [25]. The chain extension did not significantly affect the Tg of the PLLA-PEG-PLLA and blends. The Tm of PEG at 66 °C [see Fig. S1 (b)] disappeared for the PLLA-PEG-PLLA suggesting good miscibility between PEG middle-blocks and PLLA end-blocks to inhibit crystallization of PEG middle-blocks. The Tm of homo-crystallites (Tm,hc) decreased and that of the stereocomplex crystallites (Tm,sc) slightly increased as the PDLA ratio increased. This indicated that crystallization of the blends with larger PDLA ratio induces the integrity of the stereocomplex crystallites and their lamella thickness.

DSC heating curves of (above) non-chain-extended and (below) chain-extended blends of various PLLA-PEG-PLLA/PDLA compositions
It was also found that the Tm,hc peaks of blends disappeared when the PDLA ratios were increased up to 30 wt% for both the non-chain-extended and chain-extended blend series. The results suggested that only the 30 wt% PDLA induced complete stereocomplexation of PLLA end-blocks. This was due to the low M.W. PDLA acting as a better stereocomplex enhancer than the high M.W. PDLA because of its easier chain diffusion and mobility for stereocomplexation [28, 29].
The non-chain-extended PLLA-PEG-PLLA had no peak of Tcc as shown in Fig. 1 (above, a). The good chain-mobility of PLLA-PEG-PLLA is due to flexible PEG middle-blocks enhancing crystallization of PLLA end-blocks during the DSC quenching process. However, the chain-extended PLLA-PEG-PLLA exhibited a Tcc peak at 73 °C in Fig. 1 (below, a). This is due to the long-chain branched structures of chain-extended PLLA-PEG-PLLA reducing its chain diffusion and mobility for crystallization [25]. Meanwhile the Tcc peak did not detect all the chain-extended blends [Fig. 1 (below, b-e)]. This indicates the low M.W. PDLA accelerated crystallization of the chain-extended blend series.
For Xc,DSC, the hc-Xc,DSC largely decreased and sc-Xc,DSC steadily increased as the PDLA ratio increased. The chain-extended blends had lower hc-Xc,DSC than the non-chain-extended blends for the same blend ratio. It has been reported that the long-chain branched structures of chain-extended PLLA suppressed its homo-crystallization [33]. However, the sc-Xc,DSC of the blends did not change significantly by chain extension for the same blend ratio.
Crystallization behavior of the samples was investigated from DSC cooling curves as illustrated in Fig. 2. The Tc and enthalpy of crystallization (ΔHc) of non-chain-extended PLLA-PEG-PLLA (105 °C and 27.7 J/g, respectively) were higher than that of the chain-extended PLLA-PEG-PLLA (91 °C and 20.1 J/g, respectively) suggested that chain extension suppressed crystallizability of homo-crystallites of PLLA-PEG-PLLA during DSC cooling scan. This supports the conclusion that branched structures of chain-extended PLLA-PEG-PLLA inhibited homo-crystallization of PLLA end-blocks as described above.

DSC cooling curves of (above) non-chain-extended and (below) chain-extended blends of various PLLA-PEG-PLLA/PDLA compositions
The Tc peaks of the scPLA from DSC cooling scan included crystallization of both the homo- and stereocomplex crystallites [29]. In this work, the 90/10 and 80/20 (w/w) blends with and without chain extension showed smaller Tc peaks (lower ΔHc) significantly different from those of PLLA-PEG-PLLA. This may be due to the ΔHc of homo-crystallization dropping considerably when the PDLA ratios were 10 and 20 wt% while little stereocomplex crystallization occurred at these PDLA ratios [18]. For both the non-chain-extended and chain-extended blend series, the Tc peaks shifted to higher temperatures and ΔHc values steadily increased as the PDLA ratio increased because of increasing content of stereocomplex crystallites. The crystallization speed of sterecomplex crystallites was faster.
Thermal decomposition
The thermal decomposition of PLLA-PEG-PLLA and blends was investigated from TG curves as shown in Fig. 3. The PLA and PEG chains showed thermal decompositions in the ranges 240–350 °C and 350–450 °C, respectively, as compared with TG curves of PLLA and PEG (see Fig. S2). The inset thermograms illustrate the initial-step of thermal decomposition. From Fig. 3 (above), it can clearly be seen that the blends showed slower thermal-decomposition than the pure PLLA-PEG-PLLA. The stronger interactions between PLLA end-blocks and PDLA chains improved its thermal stability.

TG curves of (above) non-chain-extended and (below) chain-extended blends of various PLLA-PEG-PLLA/PDLA compositions
The chain-extended blend series showed slower thermal decomposition than the non-chain-extended blend series. It can be seen that the blends with and without chain extension started weight loss at approximately 270 °C and 240 °C, respectively. Joncryl® has been used to maintain M.W. or build high M.W. of PLA by formation of long-chain branching molecules during the melt process [35].
Derivative TG (DTG) thermograms in Fig. 4 were used to provide more data on thermal decomposition of the samples. Peaks of temperature of maximum decomposition-rate (Td, max) were detected. The Td, max peaks of PLA chains were in range 292–318 °C for the non-chain-extended blend series [Fig. 4 (above)] and significantly increased with the PDLA ratio. This supports the conclusion that PLA stereocomplexation improved thermal stability of the blends. In addition, the Td, max peaks of PEG blocks of the non-chain-extended blends (409–414 °C) were also higher than the pure PLLA-PEG-PLLA (406 °C). However, the Td, max peaks of chain-extended PLA chains slightly increased from 302 °C to 305–308 °C when the PDLA was blended. The Td, max peaks of PEG blocks of the blends with chain extension were in range 412–414 °C.

DTG curves of (above) non-chain-extended and (below) chain-extended blends of various PLLA-PEG-PLLA/PDLA compositions
Crystalline structures
Figure 5 shows XRD patterns of film samples. For the non-chain-extended blend films in Fig. 5 (above), the PLLA-PEG-PLLA film only had a diffraction peak at 17° (hc peak), attributed to the homo-crystallites of PLLA [34]. Usually the compressed PLLA films were completely amorphous. Therefore the crystallization of the PLLA end-blocks was improved by increasing their chain-mobility due to the flexible PEG middle-blocks [25]. For all the non-chain-extended blend films, the diffraction peaks assigned to homo-crystallites disappeared, and the diffraction peaks at 12°, 21° and 24° (sc peaks) ascribed to stereocomplex crystallites [34] were the only peaks observed. As PDLA ratio increased, the intensity of these diffraction peaks tended to increase. The results indicate that only stereocomplex crystallites formed in the blend films. This can be explained by external compression-forces enhancing stereocomplexation of the blend films [36]. The chain-extended PLLA-PEG-PLLA film in Fig. 5 (below) was completely amorphous. The latter film did not have any diffraction peaks. Only sc peaks of stereocomplex crystallites were detected for all the chain-extended blend films. These diffraction peaks of stereocomplex crystallites more obvious as the PDLA ratio increased.

XRD patterns of (above) non-chain-extended and (below) chain-extended blend films of various PLLA-PEG-PLLA/PDLA compositions
The hc-Xc,XRD and sc-Xc,XRD of the film samples were calculated from Eqs. (3) and (4), respectively. The non-chain-extended PLLA-PEG-PLLA film had only a hc peak (hc-Xc,XRD = 7.5%) while all the blend films had only sc peaks. The sc-Xc,XRD of the film samples were compared in Fig. 6. It can be seen that the sc-Xc,XRD of the blend films increased as the PDLA ratio increased. The sc-Xc,XRD values of the chain-extended blend film type were larger than the non-chain-extended blend film type for the same blend ratio.

sc-Xc,XRD of ( ) non-chain-extended and (
) non-chain-extended and ( ) chain-extended blend films of various PLLA-PEG-PLLA/PDLA compositions
) chain-extended blend films of various PLLA-PEG-PLLA/PDLA compositions
In previous work [32], the scPLA films of high M.W. PLLA/high M.W. PDLA blends with and without chain extension were prepared by melt blending before compression molding. The chain extension of stereocomplex PLLA/PDLA films suppressed stereocomplexation due to their rigid long-chain branching structures with high Tgs (54–55 °C) inhibiting crystallization. However, the chain-extended PLLA-PEG-PLLA/PDLA films in this work showed better stereocomplexation than the non-chain-extended PLLA-PEG-PLLA/PDLA films. This may be explained by the flexible long-chain branching structures of PLLA-PEG-PLLA-based scPLA with low Tgs (31–33 °C) enhancing crystallization.
Tensile properties
Figure 7 shows selected tensile curves of the film samples, except the 60/40 (w/w) PLLA-PEG-PLLA/PDLA blend film without chain extension because it was highly brittle. The inset tensile-curves are also presented for clarity. The blend films exhibited a yield point except for the chain-extended 60/40 blend film indicating more flexibility of these blend films.

Tensile curves of (above) non-chain-extended and (below) chain-extended blend films of various PLLA-PEG-PLLA/PDLA compositions
The averaged tensile results are compared in Fig. 8. For the non-chain-extended blend films, the stress at break of the films increased until the PDLA ratio was increased up to 20 wt%. The stereocomplex crystallites linked between the PLLA-PEG-PLLA chains acted as cross-linkers to enhance stress at break of the blend films [26, 27]. However the stress at break of the blend films dramatically dropped when the PDLA ratio was 30 wt%. The latter films became brittle because they contained a larger fraction of low M.W. PDLA. The strain at break of non-chain-extended PLLA-PEG-PLLA film increased from 33% to 148% as the PDLA ratio was 10 wt% due to the plasticizing effect of PEG middle-blocks for the film with low crystallinity (sc-Xc,XRD = 3.4%). However, the strain at break largely dropped to 10% when the PDLA ratio was 20 wt%. Brittleness was observed in the latter film.

Tensile properties of ( ) non-chain-extended and (
) non-chain-extended and ( ) chain-extended blend films of various PLLA-PEG-PLLA/PDLA compositions (
) chain-extended blend films of various PLLA-PEG-PLLA/PDLA compositions ( = could not determine)
= could not determine)
The strain at break of the PLLA-PEG-PLLA film was largely improved with chain extension. This is due to the long-chain branching structures suppressing the homo-crystallization of the PLLA end-blocks and enhancing the plasticizing effect of the PEG middle-blocks [25]. The chain-extended blend films showed better stress and strain at break than the non-chain-extended blend films for the same blend ratio. The results may be due to the longer chains of chain-extended blend films increased tie-chain density between stereocomplex crystallites [37].
In addition, the stress and strain at break of the chain-extended blend films decreased with increasing PDLA ratio. The larger fraction of low M.W. PDLA made the film brittle. The larger PDLA ratio induced higher sc-Xc,XRD that also reduced the plasticizing effect of flexible PEG middle-blocks. The Young’s moduli of blend films with and without chain extension were in ranges 705–846 MPa and 490–714 MPa, respectively.
Thermo-mechanical properties
Changes of storage modulus as a function of temperature from DMA analysis have been used to investigate the heat-resistant properties of scPLA [38, 39]. The storage modulus of amorphous PLLA with poor heat-resistance largely dropped in the Tg region before rising up again due to cold crystallization during the DMA heating scan [40]. However high crystallinity PLLA with high heat-resistance maintained its stiffness in the Tg region [8].
Figure 9 shows storage modulus of film samples from DMA. For the non-chain-extended films in Fig. 9 (above), the 100/0 and 90/10 (w/w) PLLA-PEG-PLLA/PDLA films exhibited a cold-crystallization effect at around 100 °C and 110 °C, respectively, during DMA heating. Therefore these films had poor heat-resistance. The storage modulus in the range 70–90 °C significantly increased with the PDLA ratio suggesting that the heat-resistance of films increased with increasing PDLA ratio because the higher PDLA ratio induced larger values of sc-Xc,XRD [see Fig. 6]. The heat-resistance of chain-extended films also improved with increasing PDLA ratio as shown in Fig. 9 (below). Their storage moduli in the temperature range 70–90 °C also increased with the PDLA ratio.

Storage modulus from DMA of (above) non-chain-extended and (below) chain-extended films of various PLLA-PEG-PLLA/PDLA compositions
Dimensional stability to heat
The film samples before and after testing of dimensional stability to heat are shown in Fig. 10. The dimensional stability of 60/40 (w/w) PLLA-PEG-PLLA/PDLA blend film was not determined due to its high brittleness. Both the PLLA-PEG-PLLA films with and without chain extension exhibited the largest film-extension [Fig. 10(a)] indicating they had poor heat-resistance. All the blend films after testing exhibited shorter film-extension than the PLLA-PEG-PLLA films. The results suggested that the blend films had better heat-resistance because they contained stereocomplex crystallites. The film extension significantly decreased as the PDLA ratio was increased from 10 to 20 wt%. This is due to increasing the sc-Xc,XRD of the blend films.

Photographs of dimensional stability to heat at 80 °C of (above) non-chain-extended and (below) chain-extended blend films with PLLA-PEG-PLLA/PDLA compositions of (a) 100/0, (b) 90/10, (c) 80/20, (d) 70/30 and (d) 60/40 (w/w) ( = could not determine)
= could not determine)
The heat resistance of the film samples was clearly compared with the dimensional stability to heat as calculated from Eq. (5). Figure 11 shows the results of dimensional stability to heat that were 26% and 31% for PLLA-PEG-PLLA films with and without chain extension, respectively. These values increased up to 56% and 53% when the PDLA ratio was 10 wt%. The results of dimensional stability to heat of blend films were in the range 83–91% for the PDLA ratios of 20–40 wt%. Thus, the heat resistances of the blend films were better than the PLLA-PEG-PLLA films, for both the non-chain-extended and chain-extended blend films. The stereocomplexation of PLA with the PDLA can improve heat resistance of the PLLA-PEG-PLLA films.

Dimensional stability to heat of ( ) non-chain-extended and (
) non-chain-extended and ( ) chain-extended blend films of various PLLA-PEG-PLLA/PDLA compositions (
) chain-extended blend films of various PLLA-PEG-PLLA/PDLA compositions ( = could not determine)
= could not determine)
Conclusions
The scPLA were prepared by melt blending the PLLA-PEG-PLLA with low M.W. PDLA in the absence or presence of chain extender. The DSC results showed that complete stereocomplexation was obtained when the PDLA ratio was increased up to 30 wt% for both the non-chain-extended and chain-extended blend series. In addition, the larger PDLA ratios of the blends induced faster crystallization upon the DSC cooling scan.
All the blend films fabricated by compression molding exhibited complete stereocomplexation as indicated by the XRD results. The sc-Xc,XRD of blend films increased steadily with the PDLA ratio. The non-chain-extended blend films became brittle when the PDLA ratio was increased up to 20 wt%. However, the chain-extension reaction can reduce film brittleness. The PDLA blending enhanced heat-resistance of both the non-chain-extended and chain-extended blend film series as seen from the results of DMA and dimensional stability to heat. High heat-resistance of blend films was obtained when the PDLA ratio was increased up to 20 wt%. In conclusion, the low M.W. PDLA has the potential to induce the stereocomplexation and heat-resistance of PLLA-PEG-PLLA/PDLA blends while the chain-extension reaction improved mechanical properties of the blend films. This work will expand the knowledge of scPLA that will provide more application potential for flexible and high heat-resistant scPLA products.
References
Rocca-Smith JR, Whyte O, Brachais C, Champion D, Piasente F, Marcuzzo E, Sensidoni A, Debeaufort F, Karbowiak T (2017) Beyond biodegradability of poly(lactic acid): physical and chemical stability in humid environments. ACS Sustain Chem Eng 5:2715–2762
Silva D, Kaduri M, Poley M, Adir O, Krinsky N, Shainsky-Rotiman J, Schroeder A (2018) Biocompatibility, biodegradation and excretion of polylactic acid (PLA) in medical implants and theranostic systems. Chem Eng J 340:9–14
Wu CS, Tsou CH (2019) Fabrication, characterization, and application of biocomposites from poly(lactic acid) with renewable rice husk as reinforcement. J Polym Res 26:44
Saghazadeh S, Rinoldi C, Schot M, Kashaf SS, Sharifi F, Jalilian E, Nuutila K, Giatsidis G, Mostafalu P, Derakhshandeh H, Yue K, Swieszkowski W, Memic A, Tamayol A, Khademhosseini A (2018) Drug delivery systems and materials for wound healing applications. Adv Drug Deliv Rev 127:138–166
Loganathan S, Jacob J, Valapa RB, Thomas S (2018) Influence of linear and branched amine functionalization in mesoporous silica on the thermal, mechanical and barrier properties of sustainable poly(lactic acid) biocomposite films. Polymer 148:149–157
Abudula T, Saeed U, Memic A, Gauthaman K, Hussain MA, Al-Turaif H (2019) Electrospun cellulose nano fibril reinforced PLA/PBS composite scaffold for vascular tissue engineering. J Polym Res 26:110
Nuzzo A, Coiai S, Carroccio SC, Dintcheva NT, Gambarotti C, Filippone G (2014) Heat-resistant fully bio-based nanocomposite blends based on poly(lactic acid). Macromol Mater Eng 299:31–40
Zhang X, Meng L, Li G, Liang N, Zhang J, Zhu Z, Wang R (2016) Effect of nucleating agents on the crystallization behavior and heat resistance of poly(L-lactide). J Appl Polym Sci 133:42999
Tsuji H (2016) Poly(lactic acid) stereocomplexes: a decade of progress. Adv Drug Deliv Rev 107:97–135
Cui C-H, Yan D-X, Pang H, Jia L-C, Xu X, Yang S, Xu J-Z, Li Z-M (2017) A high heat-resistance bioplastic foam with efficient electromagnetic interference shielding. Chem Eng J 323:29–36
Pan G, Xu H, Mu B, Ma B, Yang J, Yang Y (2017) Complete stereo-complexation of enantiomeric polylactides for scalable continuous production. Chem Eng J 328:759–767
El-Khodary E, Fukui Y, Yamamoto M, Yamane H (2017) Effect of the melt-mixing condition on the physical property of poly(L-lactic acid)/poly(D-lactic acid) blends. J Appl Polym Sci 134:45489
Shi X, Jing Z, Zhang G (2018) Influence of PLA stereocomplex crystals and thermal treatment temperature on the rheology and crystallization behavior of asymmetric poly(L-lactide)/poly(D-lactide) blends. J Polym Res 25:71
Cui L, Zhang R, Wang Y, Zhang C, Guo Y (2017) Effect of plasticizer poly(ethylene glycol) on the crystallization properties of stereocomplex-type poly(lactic acid). Wuhan Univ J Natur Sci 22:420–428
Srithep Y, Pholharn D (2017) Plasticizer effect on melt blending of polylactide stereocomplex. e-polymers 17:409–416
Pakkethati K, Baimark Y (2017) Plasticization of biodegradable stereocomplex polylactides with poly(propylene glycol). Polym Sci Series A 59:124–132
Bai H, Deng S, Bai D, Zhang Q, Fu Q (2017) Recent advances in processing of stereocomplex-type polylactide. Macromol Rapid Commun 38:1700454
Liu Y, Shao J, Sun J, Bian X, Feng L, Xiang S, Sun B, Chen Z, Li G, Chen X (2014) Improved mechanical and thermal properties of PLLA by solvent blending with PDLA-b-PEG-b-PDLA. Polym Degrad Stab 101:10–17
Tacha S, Saelee T, Khotasen W, Punyodom W, Molloy R, Worajittiphon P, Meepowpan P, Manokruang K (2015) Stereocomplexation of PLL/PDL-PEG-PDL blends: effects of blend morphology on film toughness. Euro Polym J 69:308–318
Jing Z, Shi X, Zhang G, Lei R (2015) Investigation of poly(lactide) stereocomplexation between linear poly(L-lactide) and PDLA-PEG-PDLA tri-block copolymer. Polym Int 64:1399–1407
Song Y, Wang D, Jiang N, Gan Z (2015) Role of PEG segment in stereocomplex crystallization for PLLA/PDLA-b-PEG-b-PDLA blends. ACS Sustain Chem Eng 3:1492–1500
Jing Z, Shi X, Zhang G (2017) Competitive stereocomplexation and homocrystallization behaviors in the poly(lactide) blends of PLLA and PDLA-PEG-PDLA with controlled block length. Polymers 9:107
Luo C, Yang CM, Xiao W, Yang J, Wang Y, Chen W, Han X (2018) Relationship between the crystallization behavior of poly(ethylene glycol) and stereocomplex crystallization of poly(L-lactic acid)/poly(D-lactic acid). Polym Int 67:313–321
Han L, Yu C, Zhou J, Shan G, Bao Y, Yun X, Dong T, Pan P (2016) Enantiomeric blends of high-molecular-weight poly(lactic acid)/poly(ethylene glycol) triblock copolymers: enhanced stereocomplexation and thermomechanical properties. Polymer 103:376–386
Baimark Y, Rungseesantivanon W, Prakymoramas N (2018) Improvement in melt flow property and flexibility of poly(L-lactide)-b-poly(ethylene glycol)-b-poly(L-lactide) by chain extension reaction for potential use as flexible bioplastics. Mater Des 154:73–80
Tsuji H, Horii F, Hyon S-H, Ikada Y (1991) Stereocomplex formation between enantiomeric poly(lactic acid)s. 2. Stereocomplex formation in concentrated solutions. Macromolecules 24:2719–2724
Tsuji H, Hyon S-H, Ikada Y (1991) Stereocomplex formation between enantiomeric poly(lactic acid)s. 3. Calorimetric studies on blend films cast from dilute solution. Macromolecules 24:5651–5656
Shao J, Xiang S, Bian X, Sun J, Li G, Chen X (2015) Remarkable melting behavior of PLA stereocomplex in linear PLLA/PDLA blends. Ind Eng Chem Res 54:2246–2253
Srisuwan Y, Baimark Y (2018) Controlling stereocomplexation, heat resistance and mechanical properties of stereocomplex polylactide films by using mixtures of low and high molecular weight poly(D-lactide)s. e-polymers 18:485–490
Xie Y, Lan X-R, Bao RY, Lei Y, Cao Z-Q, Yang M-B, Yang W, Wang Y-B (2018) High-performance porous polylactide stereocomplex crystallite scaffolds prepared by solution blending and salt leaching. Mater Sci Eng C 90:602–609
Pan G, Xu H, Ma B, Wizi J, Yang Y (2018) Polylactide fibers with enhanced hydrolytic and thermal stability via complete stereo-complexation of poly(L-lactide) with high molecular weight of 600000 and lower-molecular-weight poly(D-lactide). J Mater Sci 53:5490–5500
Baimark Y, Kittipoom S (2018) Influence of chain-extension reaction on stereocomplexation, mechanical properties and heat resistance of compressed stereocomplex-polylactide bioplastic films. Polymers 10:1218
Corre Y-M, Maazouz A, Reignier J, Duchet J (2014) Influence of the chain extension on the crystallization behavior of polylactide. Polym Eng Sci 54:616–625
Lee S, Kimoto M, Tanaka M, Tsuji H, Nishino T (2018) Crystal modulus of poly(lactic acid), and their stereocomplex. Polymer 138:124–131
Ghalia MA, Dahman Y (2017) Biodegradable poly(lactic acid)-based scaffolds: synthesis and biomedical applications. J Polym Res 24:74
Cui L, Wang Y, Guo Y, Liu Y, Zhao J, Zhang C, Zhu P (2018) Effects of temperature and external force on the stereocomplex crystallization in poly(lactic acid) blends. Adv Polym Technol 37:21743
Tsuji H, Ikada Y (1999) Stereocomplex formation between enantiomeric poly(lactic acid)s. XI Mechanical properties and morphology of solution-cast films Polymer 40:6699–6708
Si W-J, An X-P, Zeng J-B, Chen Y-K, Wang Y-Z (2017) Fully bio-based, highly toughened and heat-resistant poly(L-lactide) ternary blends via dynamic vulcanization with poly(D-lactide) and unsaturated bioelastomer. Sci China Mater 60:1008–1022
Masutani K, Kobayashi K, Kimura Y, Lee CW (2018) Properties of stereo multi-block polylactides obtained by chain-extension of stereo tri-block polylactides consisting of poly(L-lactide) and poly(D-lactide). J Polym Res 25:74
Vadori R, Mohanty AK, Misra M (2013) The effect of mold temperature on the performance of injection molded poly(lactic acid)-based bioplastic. Macromol Mater Eng 298:981–990
Acknowledgements
This work was supported by Mahasarakham University (Grant no. 6105029) and The Center of Excellence for Innovation in Chemistry (PERCH-CIC), Office of the Higher Education Commission, Ministry of Education, Thailand.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 149 kb)
Rights and permissions
About this article

Cite this article
Baimark, Y., Pasee, S., Rungseesantivanon, W. et al. Flexible and high heat-resistant stereocomplex PLLA-PEG-PLLA/PDLA blends prepared by melt process: effect of chain extension. J Polym Res 26, 218 (2019). https://doi.org/10.1007/s10965-019-1881-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-019-1881-7