
Abstract
The peculiarities of controlled copolymerization of acrylonitrile with methyl acrylate and dimethyl itaconate in the presence of copper-based catalytic system were investigated. It was shown that the polymerization proceeds in a controlled mode in accordance with ARGET ATRP mechanism. The increase of molecular weights in a strict agreement with theoretically predicted values is observed. The formation of copolymers was confirmed by NMR and MALDI TOF MS analysis. The introduction of mentioned monomers to acrylonitrile results in slight decrease of the polymerization rate. The performed calorimetric investigations showed the smoothing of exothermic effect of the oxidative stabilization of formed copolymers.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Reversible-deactivation radical polymerization (RDRP) is a versatile tool for producing of well-defined polymers with valuable properties [1,2,3,4,5,6]. It opens wide opportunities for obtaining narrow-dispersed homopolymers as well as various copolymers of random, block, graft or gradient structures. Among the general RDRP techniques the most important one is the Atom Transfer Radical Polymerization (ATRP) [6,7,8,9]. In ATRP, control over macromolecular structure is achieved using transition metal complexes. In a lower oxidation state, the transition metal catalyst activates alkyl halide generating propagating radicals and the transition metal complex in the higher oxidation state which further deactivates the propagation radical (Scheme 1). One of the main advantages of ATRP is its catalytic nature. Modern ATRP techniques such as ARGET [10], SARA [11] or ICAR [12] allow obtaining well-defined polymers using extremely low amounts of catalyst (less than 100 ppm). The possibility of conducting polymerization using low catalyst amounts is achieved due to the introduction of special reducing agents which regenerate transition metal complex to lower oxidation state. Low catalyst concentration significantly facilitates the purification of polymers and allows its use in applications sensitive to metal traces such as medicine or electronics.

The general scheme of ICAR/ARGET ATRP process
One of such permissive applications of modern ATRP techniques is the synthesis of acrylonitrile (AN) copolymers suitable for production of carbon fibers [7, 13, 14]. It is considered that the most suitable precursor polymer for producing high strength carbon fibers should have narrow molecular weight distribution (MWD) and predetermined molecular weight (MW). Narrow MWD refers to consistency of the polymer chain lengths and assists the easy dissolution of the polymer due to elimination of larger, less soluble, high MW fractions [14].
Polyacrylonitrile (PAN) homopolymer is practically not used in carbon fiber production due to its limited drawability and extremely high reactivity of nitrile groups during stabilization process. Generally, acrylonitrile copolymers containing at least 90% of AN are used for carbon fibers production [15]. Derivatives of acrylic, methacrylic and itaconic acids are used for modification of polymers. The incorporation of such monomers into polymer structure results in lowering the temperature of oxidative stabilization and improves drawability of carbon fiber precursor. Such copolymers are generally synthesized by conventional radical polymerization which results in polymers with broad MWD and uncontrolled molecular weights [16,17,18,19,20].
The application of modern methods of controlled radical polymerization for producing well-defined acrylonitrile copolymers is considered as an effective tool for obtaining high-quality carbon fiber precursors. It should be mentioned that ATRP technique was successfully applied for the synthesis of narrow-dispersed PAN homopolymers [21,22,23,24] making favorable prerequisites for successful copolymerization. Among the developed ATRP techniques, the AGET ATRP seems the most suitable one for production of required acrylonitrile copolymers due to its high efficiency and low concentrations of used catalyst [10, 25]. The investigation of possibility of obtaining well-defined copolymers of AN with methyl acrylate (MA) and dimethyl itaconate (DMI) suitable for production of carbon fibers via AGET ATRP investigated in the present work seems a challenging and actual task.
Experimental
Materials
Acrylonitrile (Sigma) was purified by distillation over calcium hydride under argon immediately before use. Dimethyl sulfoxide (DMSO) and dimethyl formamide (DMF) were dried over sodium hydroxide, distilled under reduced pressure, redried over the calcined 4-A zeolite, and then again distilled. Copper (I) bromide, carbon tetrachloride, lithium bromide, glucose, and ascorbic acid (Aldrich) were used without preliminary purification. The TPMA ligand was obtained according to the known procedure [26].
Polymerization
The determined amounts of TPMA, copper(I) bromide, glucose, or ascorbic acid (as activators) were placed in a Schlenk flask equipped with a magnetic stirrer. The flask was degassed three times and filled with argon. The calculated amounts of argon flushed DMSO, acrylonitrile (4 M solution, 15.5 wt.%), carbon tetrachloride were then added. The resulting mixture was dispensed into prepared ampoules. The ampoules were degassed by three freeze-pump-thaw cycles, sealed and placed in a thermostat for a predetermined time. The polymerization was stopped by freezing the ampoule in liquid nitrogen. The polymerization product was dissolved in dimethyl formamide and precipitated in distilled water. The polymer obtained by filtration was dried to constant weight at 70 °C under reduced pressure.
Polymer characterization
The molecular-weight characteristics of copolymers were determined by gel-permeation chromatography on a Knauer system equipped with a linear column with the exclusion limit of 2 × 106 (Phenomenex, Nucleogel GPCM-10, USA) at 40°С. The detector was an RI Detector K-2301 differential refractometer and the eluent was DMF containing 10 mmol LiBr. The instrument was calibrated using.
narrow-dispersion PMMA-based standards (Polymer Standards Service) ranging from 2400 to 970,000 g/mol. The molecular weight of PAN was calculated from a universal calibration curve and the Mark–Kuhn–Houwink equation using the coefficients for PAN and PMMA known from the literature [27]. The calculation formula was as follows:
The calorimetric studies were performed using differential scanning calorimetry (DSC) on a Setaram DSC 131 instrument. Samples of ca. 1 mg were scanned at a rate of 10 °C/min. MALDI TOF measurements were conducted using Bruker Microflex LT mass spectrometer in linear mode. (4-hydroxybenzilydene)malonitrile was used as a matrix.
Results and discussion
The copolymerization of AN with MA and DMI was conducted using a CuBr/CCl4/TPMA/glucose catalytic system successfully applied for homopolymerization of acrylonitrile earlier [23]. The choice of DMSO as a solvent was determined by good solubility of AN copolymers and its tolerance towards catalytic system resulting in higher polymer yields in comparison to DMF [28]. Itaconic acid is commonly used as a modification monomer for obtaining carbon fiber precursor [17]. A reactivity of acid group towards copper amine complexes results in catalyst decomposition and quenching of polymerization. The use of dimethyl itaconate instead of itaconic acid allowed us to solve this problem and to obtain copolymers with high yield.
Copolymerization of acrylonitrile with methyl acrylate and dimethyl itaconate using glucose as reducing agent
To reveal the optimal conditions for obtaining copolymers of AN with MA and DMI with desired molecular weights and low polydisperstity the polymerizations were conducted at 40 and 60 °C at various concentrations of monomers and initiator. The Fig. 1 depicts the dependences of conversion of monomers on time.

The dependences of monomer conversion on time for AN/MA (1, 2) and AN/MA/DMI (3, 4) copolymerization in DMSO at 40°С (1, 3) or 60°С (2, 4). [AN] = 4.0 М. [AN]:[CCl4]:[CuBr]:[glucose]:[TPMA] = 900:1:0,2:2:2,2
The analysis of the obtained results allows us to conclude that the proposed catalytic system based on copper(I)/TPMA complex is capable to conduct copolymerization of AN with MA even at 40 °C, but the polymerization proceeds very slow. The monomer conversion did not exceed 40% in 100 h making this method unacceptable for the practical application. The increase of the polymerization temperature up to 60 °C resulted in significant increase of its rate and the polymer yield. Over 80% of conversion was achieved after 75 h of polymerization. The comparison of the obtained results with the earlier published data on AN homopolymerization [23] indicates that introduction of MA into the polymerization media leads to slight decrease of conversion for the same time (51% versus 66% in 36 h). The introduction of the third monomer, DMI into polymerization media results in the further retardation of the process. In spite of the mentioned retardation the terpolymerization at 60 °C proceeds up to 80% in 100 h making this method acceptable for obtaining polymers with high yield. In case of the terpolymerization at 40 °C the monomer conversion is also rather low.
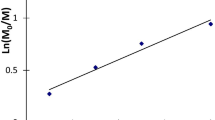
The depicted on Fig. 2 dependences of Mn and Mw/Mn values on monomer conversion are typical for ATRP processes. A linear increase of Mn with conversion is observed. It is noteworthy that the obtained Mn values are in a good agreement with theoretically calculated ones, although a slight deviation towards higher values is observed. There is no significant difference between the Mn values obtained for AN-MA and AN-MA-DMI copolymers. The formed copolymers are characterized by unimodal molecular weight distribution. The Mw/Mn values are typically 1.3–1.5 and slight higher than that of PAN homopolymer obtained in the same conditions (Mw/Mn < 1.37) [23]. The temperature of polymerization has no influence on Mw/Mn values.

The dependences of molecular weight (Mn, 1, 3) and polydispersity indices (Mw/Mn, 2, 4) of AN/MA (1, 2) and AN/MA/DMI (3, 4) copolymers obtained in DMSO at 40°С (А) or 60°С (В). [AN] = 4.0 М. [AN]:[CCl4]:[CuBr]:[glucose]:[TPMA] = 900:1:0,2:2:2,2. The straight line – theoretically calculated Mn values
Thus we can conclude that the proposed system is suitable for obtaining well-defined AN-based copolymers with AN and DMI. As the molecular weight of the obtained copolymer has a great influence on the possibility of its use as a carbon fiber precursor we tried to synthesize samples with higher molecular weights. The variation of monomer to initiator ratio in ATRP process allows to control molecular weight of the polymers and to obtain well-defined samples. Conduction of polymerization at monomer to initiator ratio equal to 1200:1 or 1900:1 resulted in formation of polymers with higher molecular weights.
The obtained data summarized in Table 1 allows us to conclude that the proposed system may be successfully applied for obtaining well-defined copolymers with high molecular weights. The copolymerization of AN with MA as well as the terpolymerization proceed up to high monomer conversions giving polymers with narrow molecular weight distribution. The molecular weights of the obtained samples gradually increase with conversion while its molecular weight distributions become narrower. The introduction of DMI as the third monomer results in slight decrease of the polymerization rate as it was observed earlier. At the same time high monomer conversions still may be achieved, but longer times are required.
Polymerization of acrylonitrile and its terpolymerization with methyl acrylate and dimethyl itaconate using ascorbic acid as reducing agent
The nature of reducing agent in AGET ATRP has a significant influence on the parameters of polymerization and the properties of the formed polymers [23]. Ascorbic acid (VC) is a well-known organic compound with high reducing activity. Being cheap and ecologically friendly biodegradable agent this compound is also commonly used in ARGET ATRP [29]. We evaluated the possibility of the use of ascorbic acid as a reducing agent in ARGET ATRP of AN and its copolymerization. The polymerization was conducted in DMSO solution at 40°, 60° and 80 °C. The Fig. 3 depicts the dependences of monomer conversion on time while the evolution of molecular weight parameters with conversion is shown on Fig. 4.

The dependences of monomer conversion on time for AN/MA (A) and AN/MA/DMI (B) copolymerization in DMSO at 40°С (1), 60°С (2), or 80°С (3). [AN] = 4.0 М. [AN]:[CCl4]:[CuBr]:[VC]:[TPMA] = 900:1:0,2:2:2,2

The dependences of molecular weight (Mn, 1, 2, 3) and polydispersity indices (Mw/Mn, 4, 5, 6) of AN/MA (a) and AN/MA/DMI (b) copolymers obtained in DMSO at 40°С (1, 4), 60°С (2, 5), or 80°С (3, 6). [AN] = 4.0 М. [AN]:[CCl4]:[CuBr]:[VC]:[TPMA] = 900:1:0,2:2:2,2. The straight line – theoretically calculated Mn values
The obtained results show that ascorbic acid can also be used as reducing agent for AGET ATRP of AN. Polymerization proceed up to high monomer conversions. Even in case of terpolymerization at 40 °C the conversion of monomers exceeds 60% in 80 h. The increase of the polymerization temperature results in the increase of the polymerization rate as for AN homopolymerization so for its copolymerization resulting in increase of monomer conversion for the predetermined period time. So, the acrylonitrile conversion in 6 h at 40 °C does not exceed 25% while its increases up to 43% and 58% respectively at 60 °C and 80 °C. The increase of the polymerization temperature has almost no influence on molecular weight distribution of samples obtained at low conversions as it was earlier observed using tin (II) 2-ethylhexanote as a reducing agent [23]. At the same time, the conduction of the process at 80 °C results in the formation of polymers with broad molecular distribution. This fact may be explained by increase of the impact of irreversible bimolecular chain termination at higher temperatures due to the increase of the activation rate (kact on Scheme 1). The higher value of kact results in increase of the concentration of propagating species. It governs the increase of polymerization rate but also leads to the higher contribution of bimolecular chain termination especially at high monomer conversions when low monomer concentration determines the decrease rate of polymerization relative to termination.
Controlled terpolymerization of acrylonitrile with methyl acrylate and dimethyl itaconate proceeds in the same manner. It should be mentioned that in the case of the use of ascorbic acid as a reducing agent the introduction of MA or DMI into polymerization media has almost no influence on monomer conversion for determined time. The rates of AN homopolymerization and its terpolymerization are almost similar.
The molecular weights of obtained samples gradually increase with conversion in a strict accordance with theoretically predicted values indicating the high control over process. At the same time some deviation is observed at high monomer conversion in case of polymerization proceeding at 80 °C. It should be mentioned that the introduction of MA and DMI into polymerization media has almost no influence on molecular weight parameters of the obtained samples. The polydispersity indices of polymers obtained at 40 and 60 °C do not exceed 1.5 indicating the high degree of control over the process.
Thus, we may conclude that both proposed AGET ATRP systems based on CuBr/TPMA catalyst may be successfully applied for copolymerization of AN with MA and DMI to obtain polymers suitable for carbon fiber production.
The determination of polymer composition
The successful formation of copolymers was confirmed by 1H NMR spectroscopy and MALDI TOF mass spectrometry. Fig. 5 represents a typical spectrum of AN/MA copolymer obtained at 82% of monomer conversion. The signal (a) at 3.05–3.25 ppm reveals the proton resonance of the CH units in the polymer backbone while the peak (b) at 1.95–2.15 is related to the proton resonance of corresponding CH2 units. Peak (c) at δ = 3.69 ppm represents the proton resonance of the methoxy unit of carbonyl, -OCH3 in MA. According to integral intensities of signals (a), (b) and (c) we can conclude that the polymer formed contains 98% of AN and 2% of MA units. In other words, the compositions of the initial monomer mixture and the polymer formed from it are almost equal.

A typical 1Н-NMR spectrum of AN/MA copolymer obtained in DMSO at 60 °C using glucose as a reducing agent
The successful formation of copolymer is also confirmed by MALDI MS spectroscopy. The mass spectrum of low-weight copolymer sample depicted on Fig. 6 is represented by several series of peaks separated either by 58 or 86 Da, corresponding to AN and MA units. These results indicate incorporation of both types of monomer units into the backbone of the polymer.

A fragment of a typical MALDI mass spectrum of AN/MA copolymer obtained in DMSO at 60 °C
The formation of terpolymers was also confirmed by means of NMR and MALDI-TOF MS. The 1H NMR spectrum of the obtained polymer is depicted on the Fig. 7. The peak (a) at = 3.1 ppm is associated with the proton resonance of the backbone CH from AN and MA units. The signal (b) at δ = 2.0–2.2 ppm reveals the proton resonance of the backbone CH2 units from AN, DMI and MA enchainment and CH2 side units of DMI. The peak (c) at δ = 3.5 ppm represents the proton resonance of methoxy of carbonyl, -OCH3, from MA and DMI while the peak (d) at δ =3.67 is associated with another methoxy -OCH3 fragment from DMI. The polymer composition calculated according to the integral intensities is represented in the Table 2. Notably, the MA content of the synthesized copolymers is slight lower than in the initial monomer mixture as the reactivity of DMI toward AN exceeds that of MA toward AN [30].

A typical 1Н-NMR spectrum of AN/MA/DMI terpolymer obtained in DMSO at 60 °C using glucose as a reducing agent
The recorded MALDI-TOF mass spectrum depicted on Fig. 8 is represented by series of lines separated by 53 and 86 Da indicating the presence of acrylonitrile and methyl acrylate units in the polymer chain. As the molecular weight of dimethyl itaconate (158) is almost equal to triple value of that for acrylonitrile (53*3 = 159) the signals from macromolecules containing DMI units are overlapped with ones bearing three AN units instead of it.

A fragment of a typical MALDI mass spectrum of AN/MA/DMI copolymer obtained in DMSO at 60 °C
Calorimetric measurements
The AN/MA copolymers and AN/MA/DMI copolymers as well as AN homopolymer were explored by means of DSC. The obtained DSC curves are depicted on Fig. 9. The obtained data indicate that glass transition temperatures as for AN homopolymer so for the obtained copolymers are almost the same and equal to 106 °C. This fact is in a good agreement with low content of comonomer units in the samples. In spite of equal glass transition temperatures the polymer behavior during oxidative stabilization is significantly different. The DSC exotherm of PAN homopolymer shows a single, very sharp and intense narrow peak with TPk = 300 °C. The sharp narrow exotherm indicates that the cyclization reaction proceeds rapidly after the initiation. The introduction of MA as a comonomer lowers the initiation temperature of the exothermic cyclization, decreases the exo-effect and results in it significant smoothing. Cyclization of terpolymer starts at lower temperature and is less exothermic. A further smoothing of the exo-peak is observed leading to the decrease of the amount of defects in carbon fiber.

The DSC curves of obtained polymers. Scan rate 10 K / min. 1 – PAN, 2-poly (AN-co-MA), 3 – poly (AN-co-MA-co-DMI)
Conclusions
The results of the performed investigations indicate that the proposed system based on copper (I) bromide in conjunction with TPMA as a supporting ligand may be successfully applied for the synthesis of well-defined acrylonitrile copolymers suitable for formation of carbon fibers. Polymerization proceeds in accordance with AGET ATRP mechanism and may be conducted using easily accessible glucose or ascorbic acid as reducing agents. Conducting polymerization in a controlled manner results in formation of copolymers with low polydispersity and desired molecular weights. The calorimetric measurements indicate that the introduction of small amounts of MA and DMI results in significant smoothing of exo-effect during oxidative stabilization making favorable conditions for the formation of carbon fibers with high mechanistic properties.
References
Lutz JF, Lehn JM, Meijer EW, Matyjaszewski K (2016). Nat Rev Mat 1:16024
Boyer C, Corrigan NA, Jung K, Nguyen D, Nguyen TK, Adnan NN, Oliver S, Shanmugam S, Yeow J (2016). Chem Rev 116:1803–1949
Anastasaki A, Nikolaou V, Nurumbetov G, Wilson P, Kempe K, Quinn JF, Davis TP, Whittaker MR, Haddleton DM (2016). Chem Rev 116:835–877
Nicolas J, Guillaneuf Y, Lefay C, Bertin D, Gigmes D, Charleux B (2013). Prog Polym Sci 38:63–235
Zaremski M, Eremeev I, Garina E, Borisova O, Korolev B (2017). J Polym Res 24:151
Poli R, Allan LEN, Shaver MP (2014). Prog Polym Sci 39:1827
Grishin DF, Grishin ID (2015). Russ Chem Rev 84:712–736
Ouchi M, Terashima T, Sawamoto M (2009). Chem Rev 109:4963–5050
Grishin ID, Kiseleva NE, Grishin DF (2015). J Polym Res 22:209
Jakubowski W, Min K, Matyjaszewski K (2006). Macromolecules 39:39–45
Konkolewicz D, Wang Y, Krys P, Zhong M, Isse AA, Gennaro A, Matyjaszewski K (2014). Polym Chem 5:4396–4417
Wang G, Lu M, Zhong M, Wu H (2012). J Polym Res 19:9782
Rahaman MSA, Ismail AF, Mustafa A (2007). Polym Degrad and Stab 92:1421–1432
Kaur J, Millington K, Smith S (2016). J Appl Polym Sci 33:43963
Park Soo-Jin. Carbon Fibers. Dordrecht: Springer. 2015. – 178 p
Li P, Shan H (1995). J Appl Polym Sci 56:877–880
Devasia R, Nair CPR, Sivadasan P, Ninan KN (2005). Polym Int 54:1110–1118
Devasia R, Nair CPR, Ninan KN (2002). Europ Polym J 38:2003–2010
Ju A, Zhang K, Luo M, Ge M (2014). J Polym Res 21:395
Jamil SNAM, Daik R, Ahmad I (2007). J Polym Res 14:379–385
Matyjaszewski K, Jo SM, Paik H, Shipp DA (1999). Macromolecules 32:6431–6438
Dong H, Tang W, Matyjaszewski K (2007). Macromolecules 40:2974–2977
Grishin ID, Kurochkina DY, Grishin DF (2017) Polym. Sci. Ser B 59:230–239
Liu XH, Zhang GB, Li BX, Bai YG, Li YS (2010) J. Polym. Sci. Part A 48:5439–5445
Pintauer T, Matyjaszewski K (2008). Chem Soc Rev 37:1087–1097
Tyeklar Z, Jacobson RR, Wei N, Murthy NN, Zubieta J, Karlin KD (1993). J Am Chem Soc 115:2611
Brandup J (1999) Polymer handbook4th edn. John Wiley and Sons, New York
Grishin ID, Kurochkina DY, Grishin DF (2015). Russ J Appl Chem 88:1275–1281
Chen H, Liu D, Ji N, Tan Z, Zong G, Qu R, Wang C (2011) J. Macromol. Sci. Part A 48:284–290
Rwei SP, Way TF, Hsu YS (2013). Polym. Degrad. and Stab. 98:2072–2080
Acknowledgments
This work was supported by Council on Grants at the President of the Russian Federation (Proj. MK-1142.2017.3) and by Russian Foundation for Basic Researches (Proj. 18-43-520016).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Grishin, I.D., Stakhi, S.A., Kurochkina, D.Y. et al. Controlled copolymerization of acrylonitrile with methyl acrylate and dimethyl itaconate via ARGET ATRP mechanism. J Polym Res 25, 261 (2018). https://doi.org/10.1007/s10965-018-1653-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-018-1653-9