
Abstract
Different types of the styrene − methyl methacrylate −acrylonitrile terpolymers were synthesized according to the recently proposed concept of the living nitroxide-mediated terpolymerization. Random azeotropic and gradient terpolymers were prepared using the living TEMPO-mediated polymerization. Block random terpolymer was obtained via the living terpolymerization initiated by macro-alkoxyamine polystyrene-SG1. In all systems, terpolymerization proceeds in the living manner even in the presence of TEMPO. Upon complete polymerization at high conversions of monomers, the molecular weight of terpolymers linearly increases with the monomer conversion. The resultant terpolymers were characterized by the methods of turbidimetric titration, GPC, DSC, and TGA, and their characteristics were compared with their non-living analogs synthesized by the conventional radical polymerization. The effect of the macromolecular structure on the thermal stability of the terpolymers was studied.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since their discovery, commercial annual production of terpolymers and multicomponent copolymers gradually increases as these compounds are being widely used in numerous valuable applications as adhesives, rubbers, functional coatings, resins, etc. In the case of the widely used conventional radical terpolymerization, the resultant products are characterized by broad MWD and chemical composition distributions at high conversions. At the present time, the current efforts of the researchers are focused on the challenging problem related to the synthesis of the products with the desired characteristics at the earlier stages of polymerization.
The powerful technique for the controlled synthesis of terpolymers is provided by the living ionic copolymerization. However, the advantages of this process are markedly restricted by the limited scope of suitable monomers. In this case, the typical products are the terpolymers of styrene, dienes and olefins, for example, styrene/isoprene/butadiene terpolymer [1]. The monomers involved in the ring-opening ionic polymerization can be also used [2,3,4]. Another serious drawback of the ionic terpolymerization process is concerned with a marked difference in the reactivities of comonomers upon polymerization. Therefore, chemically homogeneous products can be obtained via the semi-batch process which involves the controlled addition of each component in the course of the polymerization [5].
Due to the recent progress in the controlled living radical polymerization, including the nitroxide-mediated radical polymerization (NMP), atom transfer radical polymerization (ATRP), and reversible addition-fragmentation chain transfer polymerization (RAFT), a new method for the preparation of well-defined terpolymers was advanced. However, in literature, only few examples illustrating this process are available. Different random, alternating, gradient, block-random and amphiphilic terpolymers were prepared by the RAFT [6,7,8], degenerative chain transfer [9], and NMP [10, 11] techniques.
Recently [12], the general concept for the nitroxide-mediated radical terpolymerization was proposed. This concept allows the controlled synthesis of azeotropic and gradient terpolymers in the styrene − MMA − acrylonitrile (AN) system using not only a labile SG1 nitroxyl-agent but also a commonly used TEMPO nitroxide.
In this work, this concept was validated by the experimental evidence. Random, gradient and block random terpolymers were synthesized using TEMPO and SG1 nitroxides, and the resultant products were characterized by the methods of GPC, turbidimetric titration, DSC, and TGA. The properties of binary copolymers are known to be controlled not only by the type of monomers and monomer feed composition but also by the chain sequence or, in other words, by the distribution of different monomer units along the polymer chain. For terpolymers, the effect of the chain sequence on the characteristics of the resultant polymers was demonstrated for the first time. To prove this concept, the properties of “living” terpolymers were studied in comparison with the properties of their analogs prepared by the conventional free radical polymerization.
Experimental
Materials
Commercially available styrene (Pure for analysis, Russia), MMA («Sigma-Aldrich»), AN («Sigma-Aldrich») were purified according to the standard procedure and distillated under the reduced pressure. Benzoyl peroxide (BPO) was recrystallized from chloroform. TEMPO (Sigma-Aldrich) was used without additional purification. Polystyrene-SG1 alkoxyamine (PS-SG1) was obtained as described elsewhere [13]. Benzene, DMF, methylene chloride, and cyclohexane (all “Pure for analysis” Russia) were used without additional purification.
TEMPO-mediated and SG1-mediated terpolymerization were carried out according to the protocol described in [12].
Synthesis of terpolymers
Synthesis of the random homogeneous terpolymer (RH) by conventional radical polymerization
BPO (55.7 mg, 5 mM) was dissolved in the mixture of styrene (3.8 ml, 5 mol %), MMA (3.4 ml, 5 mol%), and AN (38.8 ml, 90 mol%). The reaction mixture was placed in the glass ampoule and degassed in vacuum by the triple freeze-pump-thaw cycle until a reduced pressure of 3 × 10−3 mmHg was attained; then, the ampoule was sealed. Polymerization was performed in a thermostat at 80°С for 25 min. The product was added to the 10-fold excess of the methylene chloride/benzene mixture and lyophilized in vacuum. 2.5127 g of the light yellow powder was obtained with a yield of 6.5%.
Synthesis of the random heterogeneous terpolymer (RHT) by the conventional radical polymerization
BPO (6.1 mg, 10 mM) was dissolved in the mixture of styrene (1.02 ml, 30 mol%), MMA (0.32 ml, 10 mol%) and AN (1.17 ml, 60 mol%). The reaction mixture was prepared as described above. Polymerization was carried out in a thermostat at 80°С for 24 h. The product was dissolved in the 10-fold excess of DMSO and then precipitated to the distilled water. The precipitate was washed twice by methanol and dried in vacuum. 2.0034 g of the light yellow powder was obtained with a yield of 95.7%.
Synthesis of the random-azeotropic terpolymer (RA) by the TEMPO-mediated radical polymerization
BPO (12.1 mg, 10 mM) and TEMPO (10.2 mg, 13 mM) were dissolved in the mixture of styrene (2.64 ml, 52 mol%), MMA (0.91 ml, 18 mol%), and AN (1.52 ml, 30 mol%). The reaction mixture was prepared as described above. Polymerization was carried out in a thermostat at 120°С. After a certain period of time, the reaction was stopped. The product was added to the 10-fold excess of benzene and lyophilized in vacuum in order to determine the monomer conversion.
Synthesis of the gradient terpolymer (GR) by the TEMPO-mediated radical polymerization
BPO (6.1 mg, 10 mM) and TEMPO (5.1 mg, 13 mM) were dissolved in the mixture of styrene (1.02 ml, 30 mol%), MMA (0.31 ml, 10 mol%), and AN (1.17 ml, 60 mol%). The reaction mixture was prepared as described above. Polymerization was carried out in a thermostat at 120°С for 72 h. The product was added to the 10-fold excess of the methylene chloride/benzene mixture and lyophilized in vacuum. 1.5778 g of the light yellow powder was obtained with a yield of 85.9%.
Synthesis of the block random terpolymer (BR) by the SG1-mediated radical polymerization
PS-SG1 (155.2 mg, 30 mM) was dissolved in the mixture of styrene (0.61 ml, 52 mol%), MMA (0.19 ml, 18 mol%), and AN (0.20 ml, 30 mol%). The reaction mixture was prepared as described above. Polymerization was carried out in a thermostat at 100°С. After a certain period of time, the reaction was stopped. The product was added to the 10-fold excess of the benzene and lyophilized in vacuum in order to determine the monomer conversion.
Methods
The RA and BR terpolymers were studied by the method of gel permeation chromatography (GPC) on a Waters GPC unit with a Waters GPC-system equipped with an isocratic solvent delivery system and a refractive index detector. THF was used as a mobile phase. The samples were analyzed using the system composed of three columns: 104, 105Ǻ and linear (New York) OH-Pak SB806-MHQ GPC column. The flow rate was 0.9 mL/min, and the sample concentration was 0.15% (w/v). The system was calibrated with monodisperse polystyrene (PS) standards.
МWD of RHT, RH and GR terpolymers was analyzed using GPC-120 PolymerLabs device. The chromatography was carried out at 50°С in DMF with LiBr (0.1%) as a mobile phase. Flow rate was 1.0 mL/min. The samples were separated using a system of 2 columns PLgel 5 μm MIXED B. The system was calibrated using monodisperse PMMA standards.
Isothermal calorimetry
The rate of terpolymerization was measured by the direct registration of the rate of heat release by the method of isothermal calorimetry on a Calvet type microcalorimeter DAK-1,1-A. The degree of conversion was calculated using the special computer software, and azeotropic terpolymerization was treated as the homopolymerization of an “effective” monomer with the following characteristics: average molecular weight is 88, average concentration is 10.8 M, and average value of polymerization heat is ΔH = −16.5 kcal/mol (as the arithmetic average for three monomers). For the sake of simplicity, the conversion upon gradient terpolymerization was calculated in the same manner.
Turbidimetric titration of terpolymers was performed in the DMF (solvent) —cyclohexane/methylene chloride (nonsolvent) (2: 1 by volume.) system. For the titration measurements, the solution was prepared by the dissolution of 4.5 mg of terpolymer in 3 ml of DMF. The optical density of the solution was measured on an Ultrospec 500/1100 spectrophotometer at the wavelength λ = 600 nm.
Differential scanning calorimetry
Glass transition temperatures were estimated by the DSC method on a TA Instrument Pyris 6 DSC Perkin Elmer upon heating from 30 to 150 °C at a scanning rate of 10 °C/min. The samples (15 mg) were accurately weighed, placed into the aluminum pans, and sealed.
TGA and DTA experiments were performed simultaneously for the same portion of the sample in the same experimental run on a Q-1500-D «MOM» derivatograph (Hungary). The specimen (200 mg) was placed in an open ceramic holder and heated at a heating rate of 5 °C/min in the atmosphere in the temperature interval of 24–800 °C. The sensitivity of the thermocouple in the DTA measurements was 250 mcV/oC.
The NMR analysis was performed on a VARIANXR-400 spectrometer at 400 MHz and at 298 K. The samples were prepared by the dissolution of the terpolymers in DMSO-d6, which was used as an internal standard.
The terpolymer composition was studied by the IR spectroscopy on a Specord-M80 spectrometer. The samples were prepared as thin films on a fluorite glass. The chemical composition was calculated using the absolute calibration according to the absorbance bands of MMA (ν = 1724 cm−1), styrene (ν = 1600 and 1492 cm−1), and AN units (ν = 2228 cm−1).
Results and discussion
Controlled synthesis of the azeotropic styrene − ММА − АN terpolymer via the TEMPO-mediated terpolymerization
According to [12], the TEMPO-mediated terpolymerization in the azeotropic system follows the decaying living scenario. In this system, the only living macromolecules are the macromolecules containing styrene-TEMPO and MMA-TEMPO terminal groups. The AN-TEMPO groups can be considered as “sleeping” groups. Therefore, upon polymerization, the macromolecules with the AN-TEMPO terminal adducts should be accumulated in the terpolymer. However, in this system, the monomer reactivity ratio suggests that the accumulated fraction of “sleeping” chains is not sufficient for the suppression of the living character of the TEMPO-mediated polymerization. The majority of macromolecules contains the living styrene-TEMPO terminal groups at the azeotropic point.
Figure 1 shows the kinetic curve for the TEMPO-mediated polymerization in the ternary azeotropic system. The terpolymerization is seen to proceed up to the conversion of 90–95%.

Kinetic curves of the nitroxide-mediated terpolymerization in the styrene − MMA − AN (52: 18: 30) system. [TEMPO] = 13 mM [TEMPO]/[BPO] =1.3 120оС (1); [PS-SG1] = 30 mM, 100 °C (2)
For the prepared terpolymers, all MWD curves are seen to be unimodal and, upon polymerization, the peak position is gradually shifted to the higher MW region (Fig. 2). Mn of the terpolymer linearly increases with conversion up to 60% and, then, deviation from the straight line is observed (Fig. 3). At medium conversions, PDI of the terpolymers approaches 1.3–1.5 and increases to 1.8 at final conversions.

The MWD curves of the azeotropic terpolymers obtained in the presence of TEMPO, 120оС, [ТЕМPО]/[BPO] =1.3

Mn versus conversion for the terpolymers obtained at different conversions in the styrene − MMA − AN (52: 18: 30) system. [TEMPO] = 13 mM [TEMPO]/[BPO] =1.3 120оС (1); [PS-SG1] =30 mM, 100оС (2). The values are PDI of terpolymers. The straight lines are the theoretical plots calculated according to the following equation Mn = m[M]*conversion/[BPO] (1) and Mn = m [M]*conversion/[PS-SG1] +5200 (2), where [M] is the average molar monomer concentration, m- average molar mass of monomer unit. Mn = 5200 for PS-SG1
This deviation from the living mechanism can be explained by the accumulation of “sleeping” macromolecules with the AN-TEMPO terminal groups. Moreover, the secondary inhibition reaction between TEMPO and MMA-radical can produce “dead” chains [14, 15]. Evidently, the probability of the formation of such macromolecules becomes significant only at high degrees of conversion.
Thus, in the azeotropic styrene − MMA − AN system, the TEMPO-mediated free radical terpolymerization commences as a living polymerization and finishes as a “decaying” process. This behavior is similar to the binary copolymerization of styrene and MMA or AN in the presence of TEMPO [16].
Synthesis of the gradient terpolymer via the TEMPO-mediated polymerization
The gradient terpolymer was synthesized via the living TEMPO-mediated terpolymerization in the styrene − MMA − AN (30: 10: 60) system. The choice of this monomer feed composition can be explained as follows: maximum changes in the instantaneous terpolymer composition should be observed upon terpolymerization [12]. Taking into account the reactivities of the above monomers, the consumption of monomers changes in the following order: styrene > MMA > AN. Since all growing macromolecules are living, changes in the composition of the surrounding monomer medium should lead to gradual changes in the terpolymer composition upon chain extension: the “head” of these macromolecules should be enriched with styrene, the central part – with the MMA units, and the “tail” part – with the AN units (Fig. 4). Hence, the resultant polymer is the gradient terpolymer.

The triangular diagram illustrating variations in the instantaneous terpolymer composition in the course of styrene − MMA − AN (30: 10: 60) terpolymerization. In our calculations, r 12 = 0.5, r 21 = 0.5, r 13 = 0.4, r 31 = 0.04, r 23 = 1.2, r 32 = 0.15 [17]. The arrow shows the direction of the compositional variations as the conversion increases from 0.1 to 0.9. (*) is the azeotropic point
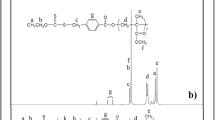
To prove the gradient structure of the living terpolymer, changes in the terpolymer composition were analyzed with increasing MW. Unfortunately, the corresponding 1H NMR-spectra are unable to confirm the gradient structure of the terpolymer. In this system, only molar ratio of styrene and MMA units can be calculated from the signals δ = 0.3–1.3 ppm (protons of α-CH3 group of MMA) and δ = 6.4–7.5 ppm (protons of the aromatic ring) (Fig. 5). All other signals are overlapped and cannot be adequately identified [18].

The 1H NMR-spectrum of the GR terpolymer obtained in the presence of TEMPO in the styrene − MMA − AN (30: 10: 60) system
Therefeore, the composition of the terpolymer was measured by the FT-IR spectroscopy. Figure 6 shows the transformation of the spectra of the living terpolymer. As follows from Fig. 6, the AN peak (2228 cm−1) becomes more pronounced, and the corresponding styrene peaks (1492 сm−1 and 700 сm−1) are seen to be reduced. Upon polymerization, the absorbance of the MMA-related peak (1724 cm−1) in the terpolymer slightly changes.

The FT − IR spectra of the terpolymers obtained in the presence of TEMPO in the styrene − MMA − AN (30: 10: 60) system
The presented experimental results qualitatively agree with the theoretical predictions according to the computational calculations within the terminal model (Table 1). However, the numerical values are seen to be slightly different. The calculations were performed according to the terminal model for the propagation reaction based on the Alfrey-Goldfiger Eqs. [19]
where Fi, is the instantaneous molar fraction of i-th monomer units in the terpolymer.
For the composition at the azeotropic point, a good agreement between experimental and calculated results was achieved. However, experimental results were somewhat different from the results of the numeral calculations for the systems enriched with the AN units. When other “modern” values of reactivity ratios, for example, from [20, 21] were used, the error in the estimation of the percentage composition is even higher. Nevertheless, both results of the numerical and spectral experiments reveal marked changes in the terpolymer composition at high conversions.
Table 1 illustrates the living character of this process. Mn of the terpolymer gradually increases up to the final conversions.
Simultaneous growth in MW and changes in the terpolymer composition confirm the gradient structure of macromolecules.
Controlled synthesis of the block-random terpolymer via the SG1-mediated terpolymerization
The block-random terpolymer was synthesized via the living terpolymerization in the azeotropic mixture in the presence of the macroinitiator PS-SG1 since this process should proceed in the “ideal” living manner up to high conversions [12]. Figure 1 shows the corresponding kinetic curves of the terpolymerization in the azeotropic system. The final conversions of monomers are close to 100%. The polymerization rates were of the same order (Fig. 1).
The “higher livingness” of the SG1-mediated terpolymerization is confirmed by the results of the GPC analysis (Fig. 7). As the conversion increases, the MWD curves are seen to be gradually shifted to the higher MW region. Mn of the terpolymer linearly increases with conversion and this behavior agrees with the theoretical dependence. For the above terpolymers, the polydispersity index changes from 1.5 at the early stages of the reaction to 1.2 at the medium conversions; at the final stage, this value is equal to 1.3. Earlier, similar results were obtained for the block-random binary copolymer P(MMA-co-S)-b-PS and terpolymer P(MMA-co-S)-b-poly(butyl acrylate) synthesized via the SG1-mediated copolymerization [22].

Molecular weight distribution curves of the block random terpolymer obtained in the PS-SG1 – styrene − MMA − AN (52: 18: 30) system. [PS-SG1] = 30 mM; 100оС
Сhemical compositional heterogeneity and thermal analysis of terpolymers
Five types of styrene − MMA—AN ternary polymers were synthesized, and chemical compositional heterogeneity of the terpolymers and their thermal stability were studied (Table 2). Three samples were living terpolymers produced at final conversions. The first random-azeotropic (RA) terpolymer was obtained by the TEMPO-mediated polymerization in the azeotropic feed mixture (52: 18: 30); the second terpolymer was prepared by the TEMPO-mediated polymerization in the feed styrene − MMA − AN mixture (30: 10: 60), and this terpolymer has the gradient (GR) structure. The third sample was the block random (BR) terpolymer obtained by the SG1-mediated terpolymerization. This terpolymer contains the PS-block and the block of the azeotropic terpolymer. Two samples were synthesized via the conventional free radical polymerization. The random heterogeneous terpolymer (RHT) was synthesized at final conversions using the feed mixture similar to that of the GR sample. This sample contains the set of macromolecules with different compositions formed at different stages of polymerization. The random homogeneous terpolymer (RH) was obtained at the low conversion (6.5%), and its composition coincides with the composition of the RHT and GR samples (Table 2).
The compositional heterogeneity of the RHT, RH and GR terpolymers was studied by the method of turbidimetric titration. At the present time, no literature data on the titration of the styrene − MMA − AN terpolymers enriched with the AN units is available. In this case, we experimentally selected the solvent–nonsolvent system as the DMF – cyclohexane/methylene chloride mixture (2/1 by volume) when, for the corresponding homopolymers, the points corresponding to the onset of clouding are different. As follows from Fig. 8, PMMA is titrated when the volume fraction of the non-solvent is γ=0.075. For PAN, this region is shifted to γ=0.8; whereas PS does not precipitate under these conditions.Footnote 1 The RH terpolymer synthesized at low conversion has a narrow clouding region at γ= 0.54–0.60 because this terpolymer is chemically homogeneous. To the contrary, the RHT terpolymer obtained at final conversions is characterized by the broad titration curve with the precipitation area ranging from γ=0.54 to 0.73. This fact correlates with the broad chemical composition distribution of the macromolecules of this polymer. For the GR terpolymer, the width of the titration curve (γ= 0.70–0.79) is similar to that of the RH terpolymer. This evidence proves the expected high compositional homogeneity of the gradient terpolymer since all its molecules grow in the same manner upon variations in the monomer feed mixture. Noteworthy is that the corresponding titration curve of the GR terpolymer is shifted to the higher values of γ since its macromolecules contain the soluble “heads” enriched with the styrene units.

Turbidimetric curves for the terpolymers with different molecular structure: random homogeneous terpolymer (RH); gradient terpolymer (GR), random heterogeneous terpolymer (RHT), and two homopolymers, PMMA and PAN. D is the optical density, γ is the volume fraction of the non-solvent (cyclohexane/CH2Cl2 = 2:1 by volume), DMF is a solvent
The most convincing evidence on the compositional homogeneity of the gradient terpolymer is the appearance of the synthesized sample. Figure 9 presents the images of the GR and RHT samples. As follows from Fig. 9, the common sample is opaque due to the microphase separation of the macromolecules with different compositions. To the contrary, the gradient terpolymer is transparent due to its homogeneous structure.

Snapshots of the samples of the GR (left) and RHT (right) terpolymers
The DSC method was used to estimate Tg for the above terpolymers. Figure 10 demonstrates that glass transition temperature is virtually independent of the terpolymer structure. Evidently, as the content of the AN units in the terpolymer increases, T g gradually decreases. Earlier, this tendency was observed for the random styrene − MMA-AN terpolymers obtained by the emulsion radical polymerization [23]. The only exception is concerned with the RHT sample which is characterized by the maximum value of T g . This behavior can be explained by the heterogeneous structure of this terpolymer, which involves stiff domains enriched with the styrene units.

The DSC curves of the terpolymers: 1− RA, 2 − GR, 3 − BR, 4 − RHT, 5 − RH
Thermal stability of the terpolymers was studied by the TGA method. Table 3 and Fig. 11 show the data on the thermal decomposition of these terpolymers. In this case, five samples with different chain structures were used: RH, RHT, GR, RA, and BR terpolymers (Table 2).

The TGA thermographs of PS, PMMA, PAN and different styrene − MMA − AN terpolymers: 1– RA, 2– BR, 3 – GR, 4 – RH, 5 – RHT
The thermographs of corresponding homopolymers were also recorded. As was expected, thermal stability of the homopolymers increases on passing from PMMA to PS and PAN.
All terpolymers show the two-step TGA curves with the basic region corresponding to the high-rate weight loss and the region corresponding to the slow-rate weight loss. The onset of the thermal decomposition of the terpolymers is close to the onset of the curve for the PS sample. For all samples under study, the temperature corresponding to the onset of the marked weight loss is equal to about 296–300 °C; for the random-azeotropic sample, this temperature slightly increases to 308 °C. For all samples, the temperature corresponding to the maximum rate of weight loss in the TGA curves remains nearly the same (347-365 °C).
However, in the high-temperature region (>420 °C), thermal stability of the terpolymers appears to be substantially different. In this temperature region, thermal stability of the terpolymers under study can be arranged in following order:
PMMA < PS < RA < BR < GR < RH ≤ RHT < PAN.
The possible reason behind this difference in the thermal stability can be attributed to the different dyad distribution of the monomer units in the macromolecules. The AN − AN dyads are known to enhance the thermal stability of polymers due to the intramolecular cyclization and formation of the thermally stable product. Therefore, the higher the content of the AN − AN dyads in the terpolymer, the higher the thermal stability of the polymer. According to the computer calculations, the content of the AN-AN dyads in the RA terpolymer is below 1%; hence, among the samples under study, the RA and BR terpolymers are characterized by the lowest thermal stability. It is interesting to compare the thermal stability of the RA and BR samples (Fig. 11, curves 1 and 2). The only difference between the block random terpolymer and the random azeotropic terpolymer is concerned with the presence of the small-sized PS block in the “head” of the macromolecules. Therefore, both samples show similar thermograms, whereas the BR terpolymer appears to be more stable in the high-temperature region because, as compared with the corresponding thermogram of the terpolymers, the thermograph of the PS sample is slightly shifted to the high-temperature region.
For three other terpolymers, namely, GR, RH and RHT, the average dyad composition appears to be the same and contains 26% of the AN − AN dyads. However, as compared with the two random samples, thermal stability of the gradient terpolymer is lower. The reason behind this behavior is still unclear.
Our results on the decomposition of the terpolymers were compared with the results of the TGA analysis [23] for three random styrene − MMA − AN terpolymers (60: 6.5: 33.5, 53.5: 13: 33.5, and 22: 29: 49) prepared by the emulsion radical polymerization. The above three terpolymers were obtained at conversions above 95% and, hence, all samples were compositionally heterogeneous. The temperatures corresponding to the onset of decomposition lie within the temperature interval of 294–311 °C, and the temperatures corresponding to the 50% weight loss were 382–386 °C. As follows from Table 3, the temperatures corresponding to the onset of decomposition fully coincide with those for the terpolymers obtained by the conventional, living, and emulsion polymerizations. However, for the terpolymers prepared via the emulsion polymerization, the temperatures corresponding to the 50% weight loss were by 15 °C higher.
This evidence suggests that thermal stability is strongly controlled by the microstructure of terpolymers as well as by the method of their synthesis.
Conclusions
Novel ternary copolymers, namely, random-azeotropic, gradient, and block random styrene − MMA − AN terpolymers were successfully synthesized via the controlled nitroxide-mediated polymerization. In this case, TEMPO and SG1 prove their potential as efficient controlling agents. For all systems, molecular weight of the synthesized terpolymers gradually increases with conversion; however, as compared with TEMPO, the potency of SG1 as a controlling agent appears to be higher.
The experiments on turbidimetric titration were used to evaluate the chemical compositional distribution of the terpolymers obtained by the living nitroxide-mediated terpolymerization as compared with the products obtained via the conventional radical polymerization. For the gradient terpolymers, the chemical compositional homogenity appears to be similar to that of the homogeneous terpolymer.
For the homogeneous terpolymers, as the content of the AN units increases, T g decreases whereas, among all terpolymers under study, the heterogeneous random sample is characterized by the maximum T g. All synthesized terpolymers shows a fair thermal stability and experience a two-step thermal decomposition with the principal high-rate weight loss at 350–400 °C and the prolong low-rate weight loss at higher temperatures. Thermal stability of the terpolymers increases in the following order: PMMA < PS < random-azeotropic terpolymer < block random terpolymer < gradient terpolymer < homogeneous random terpolymer ≤ heterogeneous random terpolymer < PAN.
Notes
RA and BR terpolymers do not precipitate under these conditions since they are enriched with the styrene units.
References
Jian Z, Tang S, Cui D (2011) Highly regio- and stereoselective terpolymerization of styrene, isoprene and butadiene with lutetium-based coordination catalyst. Macromolecules 44:7675–7981
Huijser S, HosseiniNejad E, Sablong R, de Jong C, Koning CE, Duchateau R (2011) Ring-opening co- and terpolymerization of an alicyclic oxirane with carboxylic acid anhydrides and CO2 in the presence of chromium porphyrinato and salen catalysts. Macromolecules 44:1132–1137
Hoogenboom R, Wiesbrock F, Leenen MAM, van der Loop M, van Nispen SFGM, Schubert US (2007) Kinetic investigations on microwave-assisted statistical terpolymerizations of 2-oxazoline monomers. Australian J Chem 60:656–661
Jeon JY, Eo SC, Varghese JK, Lee BY (2014) Copolymerization and terpolymerization of carbon dioxide/ propylene oxide/phthalic anhydride using a (salen)Co(III) complex tethering four quaternary ammonium salts. Beilstein J Org Chem 10:1787–1795
Gyor M, Kennedy JP (1984) Quasiliving carbocationic polymerization XVI Forced ideal terpolymerization of styrene-α-methylstyrene-isobutylene. J Macromol Sci-Chem A21:1339–1354
Hu Z, Zhang Z (2006) “Gradient” polymer prepared by complex-radical terpolymerization of styrene, maleic anhydride, and N-vinyl pyrrolidone via ç-ray irradiation by use of a RAFT process: synthesis, mechanism, and characterization. Macromolecules 39:1384–1390
Krivorotova T, Grigelis R, Jonikaitė J, Makuška R (2011) Synthesis of anionic amphiphilic molecular brushes by conventional free-radical and raft terpolymerizations. Chem Aust 22:248–254
Rixens B, Severac R, Boutevin B, Lacroix-Desmazes P (2006) Synthesis of phosphonated copolymers with tailored architecture by reversible addition-fragmentation chain transfer polymerization (RAFT). J Polym Sci: Part A Polym Chem 44:13–24
Boyer C, Ameduri B, Boutevin B, Dolbier WR, Winter R, Gard G (2008) Radical terpolymerization of 1,1,2-trifluoro-2-pentafluorosulfanylethylene and pentafluorosulfanylethylene in the presence of vinylidene fluoride and hexafluoropropylene by iodine transfer polymerization. Macromolecules 41:1254–1263
Butz S, Baethge H, Schmidt-Naake G (1999) N-oxyl mediated free radical donor-acceptor co- and terpolymerization of styrene, cyclic maleimide monomers and n-butyl methacrylate. Angew Makromol Chem 270:42–48
Lessard BH, Jee E, Ling Y, Marić M (2012) Fluorescent, thermoresponsive oligo(ethylene glycol) methacrylate/ 9-(4-Vinylbenzyl)-9H-carbazole copolymers designed with multiple LCSTs via nitroxide mediated controlled radical polymerization. Macromolecules 45:1879–1891
Zaremski MY, Plutalova AV, Eremeev I (2016) Living nitroxide-mediated radical terpolymerization. General concept and synthetic possibilities. Macromol Theor Simul 25:413–429
Laruelle G, Francёois J, Billon L (2004) Self-assembly in water of poly(acrylic acid)-based diblock copolymers synthesized by nitroxide-mediated polymerization. Macromol Rapid Commun 25:1839–1844
Zaremski MY, Zhaksylykov AB, Orlova AP, Garina ES, Badun GA, Lachinov MB, Golubev VB (2005) Reversible and irreversible inhibition in free-radical polymerization of methyl methacrylate mediated by 2,2,6,6-tetramethyl-1-piperidinyloxy. Polymer Science Ser А 47:526–535
Ananchenko GS, Souaille M, Fischer H, Le Mercier C, Tordo P (2002) Decomposition of model alkoxyamines in simple and polymerizing systems. II. Diastereomeric N-(2- methylpropyl)-N-(1-diethyl-phosphono-2,2-dimethylpropyl)-aminoxyl-based compounds. J Polym Sci: Pt A: Polym Chem 40:3264–3283
Zaremski MY, Plutalova AV, Lachinov MB, Golubev VB (2000) A concept for quasiliving nitroxide-mediated radical copolymerization. Macromolecules 33:4365–4372
Hocking MB, Klimchuk KA (1996) A refinement of the terpolymer equation and its simple extension to two- and four-component systems. J Polym Sci: Pt A Polym Chem 34:2481–2497
Brar AS, Hekmatyar SK (1999) Microstructure determination of the acrylonitrile–styrene–methyl methacrylate terpolymers by NMR spectroscopy. J Appl Polym Sci 74:3026–3032
Alfrey T, Goldfinger G (1944) Copolymerization of systems containing three components. J Chem Phys 14:115–116
Khesareh R, McManus NT, Penlidis A (2006) High temperature bulk copolymerization of methyl methacrylate and acrylonitrile. I Reactivity ratio estimation J Appl Polym Sci 100:843–851
Willemse RXE, van Herk AM (2006) Copolymerization kinetics of methyl methacrylate-styrene obtained by PLP-MALDI-ToF-MS. J Am Chem Soc 128:4471–4480
Nicolas J, Dire C, Mueller L, Belleney J, Charleux B, Marque SRA, Bertin D, Magnet S, Couvreur L (2006) Living character of polymer chains prepared via nitroxide-mediated controlled free-radical polymerization of methyl methacrylate in the presence of a small amount of styrene at low temperature. Macromolecules 39:8274–8282
Li D, Gao J, An Q, Liu G, Yang L (2001) Thermal analysis of poly(AN-co-St) and poly(AN-St-MMA). J Therm Anal Cal 63:69–74
Acknowledgements
This work was supported by the Russian Science Foundation (grant 14-13-00683).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zaremski, M., Eremeev, I., Garina, E. et al. Controlled synthesis of random, block-random and gradient styrene, methyl methacrylate and acrylonitrile Terpolymers via Nitroxide-mediated free radical polymerization. J Polym Res 24, 151 (2017). https://doi.org/10.1007/s10965-017-1303-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-017-1303-7