
Abstract
The curing kinetics of epoxy system modified with Poly(ε-caprolactone)-block-Polystyrene (PCL-b-PS) diblock copolymer was investigated by differential scanning calorimetry(DSC). PCL-b-PS was synthesized Via the combination of ROP and ATRP, then incorporated into epoxy to access the nanostructured thermosets. The results of TEM, SAXS and DSC demonstrated the occurrence of Reaction-induced Micro-phase Separation. Kinetic studies showed that the PCL-b-PS block copolymer delayed the curing reactions of epoxy system. The occurrence of Reaction-induced Micro-phase Separation had no significant effect on the total heat of reaction ∆H and the total activation energy, but resulted in a higher activation energy at the beginning of curing. The increase in activation energy at the initial stage of curing was related to the size and distribution of the dispersed phase. It is expected that the investigation of cure kinetics of EP/PCL-b-PS could provide more theoretical basis for the preparation of block copolymer modified epoxy resin.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Epoxy is one of the most widely used thermosets, with excellent physical and mechanical properties, chemical resistance, electrical insulation properties, heat resistance and bonding properties [1, 2]. As the epoxy resin and its cured system has a series of valuable properties, can be used as coatings, adhesives and fiber reinforced composite matrix resin. However, due to the high cross-linking density, the epoxy resin has a low fracture toughness. In most cases, epoxy resins is modified to compensate for the shortcomings, such as the incorporation of soft materials (eg. Liquid rubber and silicone) through blending.
The understanding of the curing kinetics of the nano-modified epoxy resin matrix can contribute to the improvement of the design and manufacture of epoxy-based composites. In general, the improvement in the performance of epoxy nanocomposites depends on the formation of crosslinked molecular networks. The curing process of the epoxy system becomes more complex after adding the modifier, which involves a series of physical and chemical changes [3]. The relationship between processing, structure and property can be clearly analyzed by studying the curing kinetics, which could determine the proper set of process parameters for the development of high-performance blends and composites with the best structural and morphological properties.
In the past decades, the blending of an epoxy resin with block copolymers consisted of an epoxy phobic block and another epoxy phylic have become a subject of some recent studies [4,5,6,7]. Compared with the traditional modification method, modification with block copolymers can allow ordered macro/nanostructures to be obtained [6,7,8,9,10]. At certain compositions, amphiphilic block copolymers spontaneously self-assemble into vesicles, spherical micelles, or wormlike micelles in thermoset resins, and these morphologies are retained with the full curing of the resins. Their excellent properties can be tailored by controlling self-assembling at the nanometer scale in the uncured and cured state. The formation of nanostructures in thermosets containing amphiphilic block copolymers has been reported to follow self-assemble [11,12,13,14] or reaction-induced [15, 16] microphase separation mechanisms. During the curing process of block copolymer modified epoxy system, the formation and immobilization of nanostructures involve a series of chemical and physical changes which are determined by the curing process and the properties of the block copolymer and epoxy. There have been some reports of the cure kinetics of epoxy/block copolymer Blends. And differential scanning calorimetry (DSC) was used to study the epoxy resin curing process, both isothermal and non-isothermal heating mode [17,18,19,20,21]. However, there are not studies on the effect of Reaction-induced micro-phase separation of block copolymer on curing kinetics of Epoxy thermosets.
In our study, poly(ε-caprolactone)-block-polystyrene (PCL-b-PS) was synthesized Via the combination of ring-opening polymerization (ROP) and atom transfer radical polymerization (ATRP) and the nanostructured thermosets modified with PCL-b-PS were obtained to investigate the effect of this block copolymer on the cure mechanism and cure kinetics of epoxy resin. For this propose, non-isothermal DSC measurement was carried out to determine the total heats of reaction released during curing for the epoxy blends modified with different contents of block copolymers. Kinetic studies showed that the PCL-b-PS block copolymer delayed the curing reactions of epoxy system. More theoretical basis for the preparation of block copolymer modified epoxy resin is expected to be provide by the investigation of cure kinetics of EP/PCL-b-PS.
Experimental
Materials
Epoxy resin (E51, Diglycidyl ether of bisphenol A-based) was purchased from Jiangsu Wuxi Resin Plant, China. 3,3′-Dichloro-4,4′-diamino diphenyl methane (MOCA), which was purchased from Changshan beier Co., China, is used as curing agent. The monomer of ε-caprolactone (ε-CL) (Aladdin, 99%) was dried over calcium hydride (CaH2) and distilled under decreased pressure prior to use. Benzyl alcohol was purchased from Chemical Reagent Factory of Kelong. Prior to use, it was distilled under decreased pressure over calcium hydride (CaH2). Stannous octanoate [Sn(Oct)2] was purchased from Aladdin Co. Styrene (St) was purchased from Chemical Reagent Factory of Kelong. It was first washed with an aqueous solution of sodium hydroxide (5 wt%) three times and washed with water until neutralization, then distilled under decreased pressure over calcium hydride (CaH2). 4-Dimethylaminopyridine (DMAP) was purchased from Nanjing Tianhua Reagent Co., China, and recrystallized in toluene at 80 °C. N,N,N′,N′,N′-pentamethyldiethylenetriamine (PMDETA), 2-bromoisobutyryl bromide and Copper(I) bromide (CuBr) were purchased from Aldrich Co.. Prior to use, CuBr was purified by stirring in glacial acetic acid overnight, filtered off, washed with ethanol, and then dried in a vacuum oven at 60 °C for 24 h. All other solvent were used as received.
Synthesis of PCL-b-PS copolymer
Synthesis of PCL
Poly(ε-caprolactone) was synthesized via the ring-opening polymerization (ROP) of ε-CL in the presence of benzyl alcohol with Sn(Oct)2 as the catalyst. Benzyl alcohol(0.488 g, 4.52 mmol), ε-CL(50 g, 438.60 mmol), Sn(Oct)2(50 mg, 1/1000 (w/w) with respect to ε-CL) were charged to a pre-dried round-bottom flask equipped with a magnetic stirrer. The flask was connected to a Schlenk line to degas via three pump-freeze-thaw cycles. Then, the flask was immersed into an oil bath, and polymerization was carried out at 120 °C for 48 h. The crude product was dissolved in a spot of dichloromethane, and the solution was dropped into a great amount of cold methanol to afford the precipitates. The collected production was dried in a vacuum oven at 40 °C for 24 h.
Synthesis of PCL-b-PS copolymer
First, mono-2-bromoisobutyryl Poly(ε-caprolactone) (PCL-Br) was prepared by following the literature method [22]. PCL-OH (46 g, 3.22 mmol), DMAP (1.179 g, 9.65 mmol) and triethylamine (TEA, 0.651 g, 6.43 mmol) were dissolved in 150 mL of CH2Cl2 and the solution was cooled to 0 °C. 2-Bromo-isobutyryl bromide (BiBB, 3.698 ml, 16.09 mmol) in 150 mL of CH2Cl2 was slowly injected dropwise. After the addition was completed, the temperature was allowed to rise to room temperature. The mixture reacted for another 24 h at room temperature under stirring. Then the solution was concentrated in a rotary evaporator, and dropped into an excessive amount of cold methanol to afford the precipitates. After recrystallized for 3 times, the production was dried in a vacuum oven at 40 °C until a constant weight.
Poly(ε-caprolactone)-b-Polystyrene diblock copolymer was synthesized by atom-transfer radical polymerization (ATRP) using mono-2-bromoisobutyryl Poly(ε-caprolactone) as a macroinitiator. Typically, mono-2-bromoisobutyryl Poly(ε-caprolactone) (PCL-Br) (15 g, 1.038 mmol)), CuBr (0.24 g, 1.667 mmol), PMDETA (144.42 mg, 0.833 mmol), and styrene (30 g, 288.462 mmol) were charged to a 250 mL pre-dried round-bottom Schlenk flask. After three pump freeze-thaw cycles, the flask was immersed into an oil bath, and polymerization was carried out at 110 °C for 16 h [23, 24]. The crude product was dissolved in a spot of tetrahydrofuran (THF), and the solution was dropped into a great amount of cold methanol to afford the precipitates. The product was dried in a vacuum oven at 40 °C until a constant weight. The process of synthesis for PCL-b-PS diblock copolymer was summarized in Scheme 1.

Synthesis of PCL-b-PS Block Copolymers
Preparation of nanostructured epoxy resin
The block copolymer PCL-b-PS was added to DGEBA and the mixtures were vigorously stirred at elevated temperature until the mixtures were homogeneous. The curing agent (MOCA) was added with vigorous stirring until homogeneous solutions were obtained again. And then, the mixture was poured into a Teflon mold. All the mixtures were cured at 150 °C for 2 h plus 180 °C for 2 h. The epoxy thermosets were prepared with contents of the PCL-b-PS diblock copolymer of 0, 20 and 40 wt%, respectively.
Measurement and characterization
Nuclear magnetic resonance spectroscopy (NMR)
The NMR measurements were carried out on a DRX-400 (Bruker Company, Germany) 400 MHz NMR spectrometer to obtain 1H-NMR spectra at 25 °C. The samples were dissolved in CDCl3.
Gel permeation chromatography (GPC)
Molecular weights of the polymers were measured on a HLC-8320 chromatograph (Tosoh, Japan) system. This apparatus was equipped with two columns (TSK gel super HM-H 6.0 *150 mm) in serials. THF was used as the eluent at the flow rate of 1.0 mL/ min at 40 °C. The molecular weights were expressed relative to polystyrene standards.
Transmission electron microscopy (TEM)
The thermosets were sliced by using an ultra-thin microtome machine equipped with a diamond blade. The sections of the samples with the thickness of 70~100 nm were placed in 200-mesh copper grids. The morphological observation was performed on a JEM 2010 high-resolution transmission electron microscope (TEM) at an accelerating voltage of 200 kV.
Small-angle X-ray scattering (SAXS)
The SAXS measurements were taken on the Xeuss 2.0 SAXS/WAXS system. Two-dimensional diffraction patters were recorded using PILATUS 3R 300 K detector. The experiments were carried out with the radiation of X-ray with the wavelength of λ = 1.54 Å at room temperature (25 °C) operating at 50 kV, 0.6 mA. The intensity profiles were output as the plot of scattering intensity (I) versus scattering vector, q = (4π/λ) sin (θ/2) (θ = scattering angle).
Thermal gravity analysis
Thermogravimetric analysis was carried out under atmospheric N2 over a temperature range of 30 °C–800 °C using a TG-209F (Netzsch) at a heating rate of 10 °C/min.
Differential scanning calorimetry(DSC)
Calorimetric studies were carried out on DSC Q200 (TA, USA) which was calibrated with high purity indium and zinc standards. Samples of about 10 mg were put in aluminum pans under nitrogen atmosphere at the heating rate of 5, 10, 15and 20 K/min, respectively. Measurements were always carried out with an empty cell as reference from 50 °C up to 350 °C.
After the curing reaction is completed, glass-transition temperatures were measured at a heating rate of 20 K/min on subsequent heating scans.
Results and discussion
Synthesis of PCL-b-PS block copolymer
Poly(ε-caprolactone)-b-Polystyrene diblock copolymer was synthesized by atom-transfer radical polymerization (ATRP) using mono-2-bromoisobutyryl Poly(ε-caprolactone) as a macroinitiator. The 1H NMR spectra of PCL-Br and the PCL-b-PS diblock copolymer were shown in Fig. 1. 1H NMR (CDCl3, δ in ppm) δ: 6.45–7.26 (m, broad, PS ArH), 4.08 (broad, CH2O of PCL), 2.30 (m, broad, OCCH2 of PCL), 1.34–1.78 (complex m, CH2 PCL and PS chain). As indicated in the 1H NMR spectrum, the simultaneous appearance of the resonance characteristic of PS and PCL protons implies that the resulting product combines the structural features of PS and PCL, which means that the PCL-b-PS block copolymer was successfully obtained.

1H NMR spectrum of PCL-b-PS block copolymer
Both the PCL and PCL-b-PS block copolymer were subjected to gel permeation chromatography (GPC) and the GPC curves are presented in Fig. 2. The PCL had a molecular weight of Mn = 14,298 g/mol with Mw/Mn = 1.27 whereas the PCL-b-PS block copolymer possessed the molecular weight of Mn = 41,975 g/mol with Mw/Mn = 1.21. The lengths of PS blocks in the block copolymer are calculated to LPS = 27,677 g/mol. The results of 1H NMR and GPC indicated that the PCL-b-PS block copolymer was successfully obtained.

GPC curves of PCL and PCL-b-PS block copolymer
Nanostructures in epoxy thermosets containing PCL-b-PS
The thermosets containing PCL-b-PS diblock copolymer were subjected to Transmission electron microscopy (TEM). Shown in Fig. 3 are the TEM images of the thermosets containing 20 and 40 wt% of the PCL-b-PS diblock copolymer. The ultrathin sections of the epoxy thermosets containing 20 and 40 wt% PCL-PS diblock copolymer are stained with RuO4 to increase the electron density contrast. For the thermosets containing 20 wt% of PCL-b -PS diblock copolymer, PS blocks were homogeneously dispersed into the continuous epoxy matrix as the spherical vesicles with ∼24 nm in diameter (Fig. 3a). For the cured blends containing 40 wt% of PCL-b-PS, morphologies transition from spherical to worm-like was observed. The worm-like structure with a diameter of ∼32 nm was observed (Fig. 3b). The nanostructures were further investigated by means of small-angle X-ray scattering (SAXS), and the SAXS profiles of the thermosets containing 0, 20 and 40 wt% of PCL-b-PS diblock copolymer are shown in Fig. 4. It is seen that the well-defined scattering peaks were observed when the content was 20 and 40 wt%, compared with the SAXS profile of pure epoxy resin, indicating that the thermosets containing PCL-b-PS are microphase-separated. According to the position of the primary scattering peaks, the average distance (according to the Bragg eq. L = 2π/Q) between the neighboring domains can be estimated to be 43.04 and 56.10 nm for the thermosetting blends containing 20 and 40 wt% PCL-b-PS, respectively. The average distance between the neighboring domains slightly increased with increasing the content of PCL-b-PS. As shown in the TEM results, the degree of aggregation of the block copolymer in the epoxy resin matrix increased, accompanied by an increase in the distance between each other. Therefore, the first peak of SAXS exhibit a blue shift exhibit a blue shift from PCL-PS-20 wt% to 40 wt%. Furthermore, for the 40 wt%, the scattering peaks of the thermosets situated at Q values of 1, 30.5 and 90.5 relative to the first-order scattering peak positions are discernible [15]. Combining with the results of TEM, these are probably the lattice scattering peaks of worm-like nanophases arranged in hexagonally packed cylinder morphology. The SAXS results were in a good agreement with the results obtained by means of TEM.

TEM micrograph of the thermosets containing 20 and 40 wt% of the PCL-b-PS diblock copolymer

SAXS profiles of the thermosets containing PCL-b-PS block copolymer
Mechanism of the formation of nanostructures.
Before curing, all the mixtures composed of DGEBA, MOCA and PCL-b-PS were homogenous and transparent, indicating that no macroscopic phase separation occurred at the scale exceeding the wavelength of visible light. It has been known that PCL was miscible with epoxy after and before curing reaction, the miscibility was ascribed to the formation of the intermolecular hydrogen bonding interaction between the aromatic amine-cross-linked epoxy and PCL [15], whereas the binary mixtures composed of PS and DGEBA displayed an upper critical solution temperature (UCST) behavior [16]. Considering the volume fraction in the epoxy blends and the results of staining, the dark areas in the TEM images should be ascribed to PS domains, while the continuous gray areas is attributed to the cross-linked epoxy.
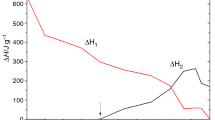
DSC curve of the neat PCL-b-PS diblock copolymer is shown in Fig. 5. DSC curve displays a sharp endothermic peak at 65 °C, which is attributed to the melting transition of PCL subchain. This observation suggests that the diblock copolymers are microphase-separated. Meanwhile, the glass transition of PS wasn’t clearly observed. Combining with the literature [24], the glass transition temperature of PS was presumed to be 60–70 °C, which overlapped with the melting temperature of PCL. During the curing process, the glass transition of PS was not observed, shown in Fig. 6. In addition, DSC curves of the thermosets containing 0, 20 and 40 wt% of the PCL-b-PS diblock copolymer after curing are shown in Fig. 7. It can be seen that the glass transition temperature (120–150 °C) of the composites slightly decreases when the content of PCL-b-PS increased, which is due to the plasticization of PCL subchains. What’s more, a transition appears at 60–70 °C, which is attributed to the glass transition of PS subchains. Therefore, the mechanism of PCL-b-PS in the formation of nanostructures in epoxy was the reaction induced microphase separation.

DSC curves of the neat PCL-b-PS diblock copolymer

DSC non-isothermal thermograms of the thermosets containing 20 and 40 wt% at a heating rate of 10 K/min

DSC curves of the thermosets containing 20 and 40 wt% of the PCL-b-PS diblock copolymer after curing reaction
Curing kinetics of epoxy nanocomposites.
DSC is one of the thermal analysis methods. Kinetics can be characterized with DSC by measuring heat generated during the curing reaction as a function of temperature and time. Before that, to elucidate the thermal stability of the epoxy resin matrix, neat EP was also examined using the thermogravimetric analysis shown in Fig. 8. The rate of mass loss showed that the initial degradation temperature is 369.7 °C, which means that no thermal degradation occured within the DSC operating temperature range (50~350 °C).

Thermal stability of neat EP
Figure 9a–c presented the DSC non-isothermal thermograms of heat flow as a function of temperature at a heating rate of 5, 10, 15 and 20 K/min respectively. These curves were basic data for the kinetic calculation. Increasing the heating rate resulted in the exothermic peaks shifting to an increasingly higher and boarder temperature range with the systematically elevated oneset temperature Toneset and peak temperature Tp.

DSC nonisothermal thermograms at various heating rates
In addition, the DSC curves were analyzed on the basis of the following assumption: the area under the curves was proportional to the conversion. So the overall reaction heat ∆H at different heating rates could be estimated by integrating the heat flow curve under the condition of removing the baseline [25]. In Fig. 10a–c, the degree of conversion (α) as a function of temperature of three systems were given. At the same time, the curve of the degree of conversion (α) as a function of temperature at the heating rate of 5 K/min of three systems were put together in Fig. 10d. The degree of conversion, α at any temperature T can be calculated by integrating the exothermic DSC peak and it is generally expressed as:
Where ∆HT is the heat of reaction of partially cured samples heat up to the temperature T and ∆H is the total heat of reaction. The degree of conversion (α) of all systems shifted to the high temperature on increasing heating rate as expected (Fig. 10), indicating that higher percentage of curing reaction occurring only at higher temperature. All curves are similar and are S-shaped curves, indicating that the curing reaction of the epoxy resin is autocatalytic. When the concentration of the block copolymer was the same, for higher heating rates, higher temperatures are required for the same conversion. On the other hand, when the heating rate was the same, a higher temperature and more time are required to achieve the same conversion rate. This fact indicated that the addition of the block copolymer retarded the progress of the curing reaction.

Degree of conversion as a function of temperature
One factor could be the dilution effect of PCL-b-PS [26, 27]. Figure 9d shows the dynamic thermograms for the neat EP system and for the mixtures containing various amounts of the PCL-b-PS copolymer at the heating rate of 5 K/min. And Table 1 shows the enthalpies of reaction from dynamic measurements at the heating rate of 5 K/min. According to the results, ∆H decreased in proportion to the copolymer content in the sample, which means that the amount of reactive groups within the same volume decreased proportionally as the block copolymer content increased. This confirms that PCL-b-PS displays a dilution effect. However, the presence of PCL-b-PS did not apparently change the reaction pathway [28].
Another factor to be considered could be related to specific interactions of the epoxy matrix with the block copolymer. This change is due to interactions between the hydroxyl groups of the growing epoxy thermoset and the PCL chain of the block copolymer. It has been reported that the intermolecular hydrogen bonding interactions between the PCL subchains and the cross-linked epoxy matrix [29]. Thus, this could be due to the fact that the OH groups which developed in the cure reactions interact with the PCL ester groups through hydrogen bonding, so decreasing the autocatalytic process [30], and therefore delaying curing.
Kissinger method
The overall reaction activation energy is quite important to better understand the curing reaction. Kissinger Method has been used to evaluate the overall reaction activation energy [25, 31, 32]. From the Kissinger equation, overall activation energy can be calculated by Eq.(2). For non-isothermal curing, the relationship between activation energy Ea, the heating rate, and the temperature Tp at which the exothermic peak has its maximum can be described as:
Where R is the gas constant, equal to 8.3144 J/(K mol), β is the heating rate, Tp is peak temperature for β, and Ea is the apparent activation. The resulting slope of linear plot of ln(β/Tp2) against Tp−1 can be used to calculated Ea.
Figure 11 shows the relationship between ln(β/Tp2) and Tp−1 for the neat systems and the mixtures containing various amounts of the PCL-b-PS copolymer. The four points correspond to the four heating rates of 5, 10, 15, and 20 K/min. The results were three excellent linear lines, indicating that the Kissinger model is suitable for the experimental data. From the slopes of these three plots, the overall activation energy for neat EP, EP/LS-20 and EP/LS-40 were found to be 58.0 kJ/mol, 56.6 kJ/mol and 58.4 kJ/mol, respectively. The slight difference between the results indicated that the occurrence of reaction induced microphase separation had little effect on the total activation energy of the epoxy curing process.

Kissinger plots for the thermosets containing PCL-b-PS block copolymer
Ozawa method
In addition,Ozawa Method has been used to determine the activation energy corresponding to different stages of cure throughout the entire conversion.
The relationship between logβ vs. T−1 for nine different degrees of conversion ranging from 0.1 to 0.9 are shown in Fig. 12a-c. The linear relationship observed in all cases indicates that the approach is applicable for this case. For the purpose of comparison, the activation energies Ea calculated for three systems are given in Fig. 12d. It can be clearly seen that with the increase in the content of the block copolymer PCL-b-PS, the activation energy of the reaction at the initial stage of curing (α< 0.7) was significantly increased. The curing process of pure EP and EP/LS-20 can be divided into three stages: α<0.3, 0.3 ≤α≤ 0.7, α> 0.7. In contrast, the system EP/LS-40 had much more higher activation energies at the initial stage of curing than that of EP and EP/LS-20 systems.

Isoconversional plots at various conversions for three systems (a-c) and Variation of Ea versus α by Ozawa method (d)
This fact could be attributed to the occurrence of reaction induced microphase separation. On the one hand, the results DSC demonstrated the occurrence of Reaction-induced Micro-phase Separation. It should be noted that the phase separation occurs mainly at the beginning of the curing reaction [33], since the movement of the polymer chains becomes very difficult at the high cross-linking density. While the movement of the molecular chain involved in the microphase separation process hinders the establishment of the epoxy matrix crosslinking network, which increased the activation energy of the curing. On the other hand, considering the results of TEM and SAXS, a more complex nanostructure was obtained in thermosetting blends containing 40 wt% PCL-b-PS as compared to thermosetting blends containing 20 wt% PCL-b-PS. In contrast, the thermosetting blends containing 40 wt% PCL-b-PS were filled with worm-like nanostructures with a large aspect ratio, which occupies a larger volume than the spherical dispersed phase formed in the thermosetting blends containing 20 wt% PCL-b-PS. The reaction-induced microphase separation occured in the system EP/LS-40 therefore had a greater hindrance to the establishment of the cross-linked network, which increased the activation energy more. Finally, after the end of reaction-induced microphase separation, the activation energy of the three systems tend to be consistent.
Conclusions
This work is an investigation of curing kinetics of epoxy system modified with PCL-b-PS diblock copolymer. The results of Transmission electron microscopy (TEM), Small-Angle X-ray Scattering (SAXS) and differential scanning calorimetry(DSC) demonstrated the occurrence of Reaction-induced Microphase Separation. The curing kinetic parameters of pure epoxy and the EP/PCL-b-PS were obtained by Kissinger method and Ozawa Method. Kinetic studies showed that the PCL-b-PS block copolymer delayed the curing reactions of epoxy system. According to the results, a clearly delay of cure kinetics has been found with the increase of block copolymer content. The occurrence of Reaction-induced Micro-phase Separation did not apparently change the reaction pathway, since there was no significant change in the overall activation energy and the ∆H decreased in proportion to the copolymer content in the sample. However, the higher activation energy at the beginning of curing was observed. The reaction-induced microphase separation occured in the system EP/LS-40 had a greater hindrance to the establishment of the cross-linked network, which increased the activation energy more. It is expected that the investigation of cure kinetics of EP/PCL-b-PS could provide more theoretical basis for the preparation of block copolymer modified epoxy resin.
References
Allaoui A et al (2002) Mechanical and electrical properties of a MWNT/epoxy composite. Compos Sci Technol 62(15):1993–1998
Lee, H. and K. Neville, Handbook of epoxy resins. 1967
Shanmugharaj AM, Ryu SH (2012) Study on the effect of aminosilane functionalized nanoclay on the curing kinetics of epoxy nanocomposites. Thermochim Acta 546(546):16–23
Dean JM, Lipic PM, Grubbs RB, Cook RF, Bates FS (2001) Micellar structure and mechanical properties of block copolymer-modified epoxies. J Polym Sci B Polym Phys 39(23):2996–3010
Grubbs RB, Dean JM, Broz ME, Bates FS (2000) Reactive block copolymers for modification of thermosetting epoxy. Macromolecules 33(26):9522–9534
Marc A. Hillmyer, et al., Self-Assembly and Polymerization of Epoxy Resin-Amphiphilic Block Copolymer Nanocomposites. J Am Chem Soc, 1997. 119(11): p. 76–77
Paul M. Lipic, Frank S. Bates, and, M.A. Hillmyer, Nanostructured Thermosets from Self-Assembled Amphiphilic Block Copolymer/Epoxy Resin Mixtures. J Am Chem Soc, 1998. 120(35): p. 8963–8970
Li Q, Zaiser M, Koutsos V (2004) Carbon nanotube/epoxy resin composites using a block copolymer as a dispersing agent. Phys Status Solidi 201(13):R89–R91
Liu J et al (2008) Nanocavitation in self-assembled amphiphilic block copolymer-modified epoxy. Macromolecules 41(20):7616–7624
Rebizant V et al (2003) Reactive Tetrablock copolymers containing Glycidyl methacrylate. Synthesis and morphology control in epoxy−amine networks. Macromolecules 36(26):9889–9896
Hillmyer MA, Lipic PM, Hajduk DA (1997) Self-assembly and polymerization of epoxy resin-amphiphilic block copolymer nanocomposites. J Am Chem Soc 119(11):76–77
Maiez-Tribut S, Pascault JP, Soulé ER, Borrajo J, Williams RJJ (2007) Nanostructured epoxies based on the self-assembly of block copolymers: a new miscible block that can be tailored to different epoxy formulations. Macromolecules 40(4):1268–1273
Ramos JA, Serrano E, Tercjak A, Salgueiro W, Goyanes S, Mondragon I (2007) PALS study of epoxy matrices: self-assembly of block copolymers and its capability for nanostructuring thermosetting systems. Phys Status Solidi 4(10):3690–3699
岸, 肇, (2014) Self-assembly phase structures and functional properties of epoxy polymer blends modified with block copolymers. 日本ゴム協会誌 = J Soc Rubber Sci Technol, Japan, 87(6): p. 213–218
Xu Z, Zheng S (2007) Reaction-induced Microphase separation in epoxy thermosets containing poly(ε-caprolactone)-block-poly(n-butyl acrylate) Diblock copolymer. Macromolecules 40(7)
Fan W, Zheng S (2008) Reaction-induced microphase separation in thermosetting blends of epoxy resin with poly(methyl methacrylate)- block -polystyrene block copolymers: effect of topologies of block copolymers on morphological structures. Polymer 49(13–14):3157–3167
Lee JY, Choi HK, Shim MJ, Kim SW (2000) Kinetic studies of an epoxy cure reaction by isothermal DSC analysis. Thermochim Acta 343(1–2):111–117
Nicolas S et al (2003) A study of epoxy-amine cure kinetics by combining Isoconversional analysis with temperature modulated DSC and dynamic Rheometry. Macromol Chem Phys 204(15):1815–1821
Roşu D, Caşcaval CN, Mustaţǎ F, Ciobanu C (2002) Cure kinetics of epoxy resins studied by non-isothermal DSC data. Thermochim Acta 383(1):119–127
Sbirrazzuoli N, Vyazovkin S (2002) Learning about epoxy cure mechanisms from isoconversional analysis of DSC data. Thermochim Acta 388(1–2):289–298
Sourour S, Kamal MR (1976) Differential scanning calorimetry of epoxy cure: isothermal cure kinetics. Thermochim Acta 14(1):41–59
And JD, Chen Y (2004) PCL star polymer, PCL-PS Heteroarm star polymer by ATRP, and Core-Carboxylated PS star polymer thereof. Macromolecules 37(10):3588–3594
Yu R, Zheng S, Li X, Wang J (2012) Reaction-induced Microphase separation in epoxy thermosets containing block copolymers composed of polystyrene and poly(ε-caprolactone): influence of copolymer architectures on formation of nanophases. Macromolecules 45(22):9155–9168
Meng F, Xu Z, Zheng S (2008) Microphase separation in thermosetting blends of epoxy resin and poly([epsilon]-caprolactone)-block. Macromolecules 41(4):1411–1420
Vyazovkin S, Burnham AK, Criado JM, Pérez-Maqueda LA, Popescu C, Sbirrazzuoli N (2011) ICTAC kinetics committee recommendations for performing kinetic computations on thermal analysis data. Thermochim Acta 520(1–2):1–19
Barone L, Carciotto S, Cicala G, Recca A (2006) Thermomechanical properties of epoxy/poly([epsilon]-caprolactone) blends. Polym Eng Sci 46(11):1576–1582
Hoppe CE, Galante MJ, Oyanguren PA, Williams RJJ, Girard-Reydet E, Pascault JP (2002) Transparent multiphasic polystyrene/epoxy blends. Polym Eng Sci 42(12):2361–2368
Martinez I, Martin MD, Eceiza A, Oyanguren P, Mondragon I (2000) Phase separation in polysulfone-modified epoxy mixtures. Relationships between curing conditions, morphology and ultimate behavior. Polymer 41(3):1027–1035
Clark JN, Daly JH, Garton A Hydrogen bonding in epoxy resin/poly(∈ヽaprolactone) blends. J Appl Polym Sci 29
Swier S, van Assche G, Vuchelen W, van Mele B (2005) Role of complex formation in the polymerization kinetics of modified epoxy−amine systems. Macromolecules 38(6):2281–2288
Augis JA, Bennett JE (1978) Calculation of the Avrami parameters for heterogeneous solid state reactions using a modification of the Kissinger method. J Therm Anal Calorim 13(2):283–292
Lee JY, Shim MJ, Kim SW (2001) Effect of modified rubber compound on the cure kinetics of DGEBA/MDA system by Kissinger and isoconversional methods. Thermochim Acta 371(1–2):45–51
Fan W, Wang L, Zheng S (2014) Double reaction-induced Microphase separation in epoxy resin containing polystyrene-block-poly(ε-caprolactone)-block-poly(n-butyl acrylate) ABC triblock copolymer. Macromolecules 43(24)
Acknowledgements
The authors would like to thank the National Natural Science Foundation of China (51273118), the Fundamental Research Funds for the Central Universities (2012017yjsy186) and the Fundamental Research Funds for the Central Universities of China (2015SCU11008) for financial support.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhang, H., Heng, Z., Chen, Y. et al. The effect of reaction-induced micro-phase separation of block copolymer on curing kinetics of epoxy thermosets. J Polym Res 25, 98 (2018). https://doi.org/10.1007/s10965-018-1498-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-018-1498-2