
Abstract
A series of soluble polyimides with high glass transition temperature (T g ) were prepared from four commercial aromatic dianhydrides (i.e., BPDA, OPDA, 6FDA and BPADA) with a rigid aromatic diamine containing tert-butyl groups (3,3′-di-tert-butylbenzidine) (1). The number-average molecular weights of them were in the range from 4.21 × 104 to 1.25 × 105 with the polydispersity indexes between 2.34 and 3.04 by means of gel permeation chromatography relative to a polystyrene standard. They were able to form transparent, flexible and tough films by polymer solution casting. The transmittance of them was higher than 90 % in a wavelength range of 400–700 nm by UV–vis measurement. The XRD results demonstrated that they were amorphous glassy polymers. Except for polyimide 1-BPADA, the T g s of the rest were higher than 330 °C, and especially the T g of polyimide 1-BPDA reached up to 375 °C. They did not show appreciable decomposition up to 500 °C under a nitrogen atmosphere, in other words, the 5 % weight losses of them were in the range from 508 to 523 °C. Most of them exhibited high tensile strength and modulus with a low elongation at break at room temperature. They are potential candidates for high performance materials.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Aromatic polyimides are a very interesting polymer family with incredibly strong mechanical properties and astoundingly heat and chemical resistant. They often replace glass and metals, such as steel, in many demanding industrial applications, including advanced composites, engineering plastics and so on [1]. Polyimides are even used in many everyday applications, such as used for the struts and chassis in some cars as well as some parts under-the-hood because they can withstand the intense heat and corrosive lubricants, fuels, and coolants cars require. They can also be used in circuit boards [2], insulation [3], fibers for protective clothing [4], composites and adhesives [5, 6]. However, the commercial applications of traditional aromatic polyimides are limited because of their poor solubility and high softening or melting temperatures [7]. Therefore, the processing is usually carried out with a poly(amic acid) intermediate, which then can be converted into the polyimide via rigorous thermal treatment or chemical treatment. Unfortunately, this process has some inherent drawbacks, such as the release of volatile byproducts (e. g., H2O) and the storage instability of poly(amic acid) [8]. In order to overcome such problems, many considerable efforts have been made to improve the solubility and melting processability of polyimides in completely imidized form, including the incorporation of flexible linkages [9–16], aliphatic moiety [17, 18], fluorine atoms and bulky pendent groups into the polymer backbone during the past decades [19–23]. Generally the incorporation of flexible linkages and aliphatic moiety into the backbones may deteriorate the inherent thermal performance of polyimides, especially result in the decreasing of T g s [9–18]. Meanwhile the introduction of fluorine atoms into the backbone may drastically increase the cost of polyimides. On the contrary, the introduction of bulky pendent groups into the backbone can not only improve the processability of polyimides through destroying the close-packing of their backbones, but also maintain the high T g of polyimides by reducing the torsional mobility of their main chains [24].
According to some previous reports, tert-butyl groups have been introduced into aromatic polyimides as a large pendent group in order to improve their solubility without sacrificing thermal stability [25–29]. However, most of them have relatively lower T g s comparing with traditional aromatic polyimides due to one or more flexible linkage existing in their repeating units. Early this century, we also synthesized some highly soluble aromatic polyimides with better thermal stability by attaching tert-butyl groups to the polyimide backbones [30, 31]. Nevertheless, there is a flexible linkage (−CH2-) still between the two benzene rings of the diamine 4,4′-methylenebis(2-tertbutylaniline) (MBTBA) resulting in a relatively lower T g s. In this paper, we first synthesized a more rigid aromatic diamine 3,3′-di-tert-butylbenzidine (1) containing tert-butyl groups but without a flexible linkage between the two benzene rings. Then a series of aromatic polyimides were prepared from diamine 1 and various commercial dianhydrides. We expect these polyimides can possess the high T g s while still keep the good solubility and thermal stability.
Experimental
Materials
2-tert-butylaniline (Wuxi TPW Pharmaceutical Technology Co. Ltd.) was purified by distillation under reduced pressure before use. m-Cresol (Sinopharm Chemical Reagent Co. Ltd.) was purified by the vacuum distillation over P2O5 before used. Pyromellitic dianhydride (PMDA, Sinopharm Chemical Reagent Co. Ltd.), 3,3′,4,4′-diphenylether tetracarboxylic dianhydride (OPDA, TCI Chemicals), 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BPDA, J&K Chemical Co. Ltd.), 3,3′,4,4′-benzophenonetetracarboxylic dianhydride (BTDA, J&K Chemical Co. Ltd.), 2,2-bis(3,4-dicarboxyphenyl)-1,1,1,3,3,3-hexafluoropropane dianhydride (6FDA, J&K Chemical Co. Ltd.) and 2,2-bis [4-(3,4-dicarboxyphenoxy)phenyl]propane dianhydride (BPADA, Shanghai Research Institute of Synthesis Resins) were recrystallized from acetic anhydride and dried under vacuum at 150 °C for 12 h before used. Isoquinoline (J&K Chemical Co. Ltd.) and other materials purchased from domestic chemical markets were used without further purification.
Measurement
1H and 13C NMR spectra were recorded on a 400 MHz Varian NMR spectrometer with DMSO-d 6 and CDCl3 as solvents. The FTIR spectra were measured with a Perkin-Elmer Fourier transform infrared spectrometer. The mechanical behaviour of the polyimide films was performed by a CMT-4104 (SANS, Shenzhen, China) tensile tester with a crosshead speed of 1 mm/min at 25 ± 2 °C. The X-ray diffraction (XRD) data were recorded on a Rigaku D/max-2200/PC X-ray diffractometer using Cu/K-α radiation with 2θ in the range from 5 to 40°. The molecular weights of the polymers were evaluated by a Perkin-Elmer Series 200 gel permeation chromatography (GPC) analyzer relative to polystyrene standards. The UV–vis spectra were obtained on Perkin-Elmer Lambda 20 spectrometer. Differential scanning calorimetry (DSC) was carried out on a PE Pyris-1 thermal analyzer under nitrogen atmosphere at a heating rate of 20 °C/min from 40 to 430 °C. Thermogravimetric analysis (TGA) was performed on a PE Pyris-7 thermal analyzer under nitrogen at a heating rate of 20 °C/min from 50 to 800 °C. The molecular weight of diamine 1 was measured with a waters Q-Tof Premier mass spectrometry.
Intermediate and monomer synthesis
2-tert-Butyl-4-iodo-phenylamine (2)
1.49 g (10 mmol) 2-tert-butylaniline, 1.50 g (18 mmol) NaHCO3 were added into 10 mL vigorously stirred deionized water in a 50 mL single-neck flask. The mixture was cooled to 0 °C and then 2.54 g (10 mmol) I2 was added into it in batches. The reaction mixture was warmed up to room temperature and kept stirring constantly at room temperature for 12 h. The reaction mixture was further diluted with additional 10 mL deionized water and extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was washed with 10 mL saturated NaHSO3 solution, 10 mL saturated NaHCO3 solution, 10 mL deionized water and at last dried with anhydrous Na2SO4 overnight. After filtration and concentration under reduce pressure, some black oil was obtained. It was purified by column chromatography to give 2.39 g yellow oil (2). Yield: 87 %; 1H NMR (400 MHz, CDCl3, δ) 7.47 (d, J = 2 Hz, 1H; Ar H), 7.29 (dd, J = 8, 2 Hz, 1H; Ar H), 6.42 (d, J = 8 Hz, 1H; Ar H), 1.39 (s, 9H; CH3); 13C NMR (101 MHz, CDCl3, δ) 170.53 (Ar C), 144.37 (Ar C), 135.21 (Ar C), 134.87 (Ar C), 119.42 (Ar C), 59.93 (Ar C), 28.87 (C), 20.49 (CH3).
3,3′-Di-tert-butylbenzidine (1)
483 mg (1.755 mmol) 2,19.7 mg (0.088 mmol) Pd(OAc)2, 282 mg (0.876 mmol) tetrabutylammonium bromide, 186 mg (1.346 mmol) K2CO3 were added to a stirring mixture of 0.875 mL i-PrOH, 3.948 mL DMF and 1.535 mL deionized water under N2 atmosphere. The reaction mixture was heated to and kept at 110 °C for 12 h under the stirring. 30 mL EtOAc was added into after the reaction mixture was cooled to room temperature. The organic layer was separated by the separatory funnel, washed with deionized water (3 × 20 mL) and dried with anhydrous Na2SO4. Some red oil was produced after the filtration and concentration under the reduce pressure. After purified by column chromatography, the crude production of 1 was obtained. Furthermore, it was purified by recrystallization in ethanol for three times and afford 132 mg light red solid 1. Yield: 51 %; M. p.: 137.02 °C (determined by means of DSC measurement); 1H NMR (400 MHz, DMSO-d 6 , δ): 7.23 (d, J = 2 Hz, 2H; Ar H), 7.08 (dd, J = 8, 2 Hz, 2H; Ar H), 6.69 (d, J = 8 Hz, 2H; Ar H), 4.70 (s, 4H; NH2), 1.38 (s, 18H); 13C NMR (101 MHz, CDCl3, δ): 143.03 (Ar C), 133.81 (Ar C), 132.42 (Ar C), 125.11 (Ar C), 125.06 (Ar C), 118.15 (Ar C), 34.34 (C), 29.61 (CH3). HRMS (ESI, m/z): [M + H]+ calcd for C20H29N2, 297.2331; found, 297.2325.
Polymer synthesis
All the polyimides were synthesized by one-pot polycondensation and a typical procedure displayed as follows (e.g., 4b 1-BPDA). A 50 mL three necked flask with a nitrogen inlet and a mechanical stirring was charged with 500 mg (1.687 mmol) 1, 15 mL m-cresol, 496.2 mg (1.687 mmol) BPDA and two drops of isoquinoline under N2 atmosphere. The mixture was stirred and smoothly heated to 85 °C until the solids dissolved completely, and further kept stirring at this temperature for 12 h. Then the temperature of the reaction mixture was processed with the following heating programs, at 100 °C for 0.5 h, 120 °C for 12 h, 150 °C for 12 h and 220 °C for 12 h. After cooling to room temperature, the resulting viscous mixture was diluted by 20 mL CHCl3 under the stirring. Finally, the transparent polyimide solution was poured slowly into 500 mL vigorously stirred methanol to afford a fiber-like precipitate. After filtration, the precipitated polyimide was washed with methanol repeatedly, collected and dried under vacuum at 100 °C for 24 h. For further purification, the polyimide 4b was reprecipitated twice from DMAc into methanol. Yield is 890 mg (95 %).
4b (1-BPDA)
Yield: 95 %. IR (solution casting film, cm−1): ν = 3,648 (−NH or H2O), 3,485 (−NH or H2O), 3,037, 2,964 (CH2-H), 1,779 (C = O), 1,721 (C = O), 1,621, 1,489, 1,369 (C-N), 1,261, 1,215, 1,100, 865, 824, 746, 701, 635, 498. 1H NMR (400 MHz, DMSO-d 6 , δ): 8.56 (m, 4H; Ar H), 8.24 (s, 2H; Ar H), 7.95 (s, 2H; Ar H), 7.78 (s, 2H; Ar H), 7.60 (s, 2H; Ar H), 1.43 (s, 18H; CH3) ppm. ELEM. ANAL. Calcd. for (C36H30N2O4)n: C, 77.96 %; H, 5.45; N, 5.05 %. Found: C, 77.18; H, 5.57; N, 5.00 %.
4d (1-OPDA)
Yield: 97 %. FTIR (solution casting film, cm−1): 3,489 (−NH or H2O), 3,067, 2,964 (CH2-H), 1,780 (C = O), 1,724 (C = O), 1,609, 1,475, 1,369 (C-N), 1,275, 1,101, 956, 866, 810, 751, 696, 634, 575, 497. 1H NMR (400 MHz, DMSO-d 6 , δ): 8.13 (s, 2H; Ar H), 7.84 (s, 2H; Ar H), 7.79–7.62 (m, 6H; Ar H), 7.49 (s, 2H; Ar H), 1.33 (s, 18H; CH3) ppm. ELEM. ANAL. Calcd. for (C36H30N2O5)n: C, 75.77; H, 5.30; N, 4.91 %. Found: C, 75.05; H, 5.36; N, 4.91 %.
4e (1-6FDA)
Yield: 98 %. FTIR (solution casting film, cm−1): 3,651 (−NH or H2O), 3,497 (−NH or H2O), 2,964 (CH2-H), 1,788 (C = O), 1,729 (C = O), 1,625, 1,488, 1,370 (C-N), 1,258, 1,193, 1,146, 1,104, 1,021, 985, 866, 815, 724, 712, 636, 572, 498. 1H NMR (400 MHz, DMSO-d 6 , δ): 8.22 (d, J = 7 Hz, 2H; Ar H), 7.98 (s, 2H; Ar H), 7.91 (d, J = 8 Hz, 2H; Ar H), 7.84 (s, 2H; Ar H), 7.67 (s, 2H; Ar H), 7.53 (s, 2H; Ar H), 1.32 (s, 18H; CH3) ppm. ELEM. ANAL. Calcd. for (C39H30F6N2O4)n: C, 66.47; H, 4.29; N, 3.98 %. Found: C, 66.41; H, 4.64; N, 3.80 %.
4f (1-BPADA)
Yield: 92 %. FTIR (solution casting film, cm−1): 3,486 (−NH or H2O), 3,037, 2,967 (CH2-H), 2,688 (CH2-H), 2,346, 1,902, 1,779 (C = O), 1,725 (C = O), 1,602, 1,480, 1,368 (C-N), 1,276, 1,239, 1,172, 1,100, 1,015, 953, 853, 752, 697, 635, 548, 498. 1H NMR (400 MHz, DMSO-d 6 , δ): 7.99 (s, 2H; Ar H), 7.81 (s, 2H; Ar H), 7.63 (s, 2H; Ar H), 7.39 (s, 8H; Ar H), 7.16 (s, 6H; Ar H), 1.72 (s, 6H; Ar H), 1.28 (s, 18H; CH3) ppm. ELEM. ANAL. Calcd. for (C51H44N2O6)n: C, 78.44; H, 5.68; N, 3.59 %. Found: C, 78.39; H, 5.82; N, 3.55 %.
Results and discussion
Monomer synthesis
The diamine monomer 1 was synthesized according to a previous report and the detailed synthetic route was outlined in Scheme 1 [32]. Firstly, an iodine substituted intermediate 2 was prepared through the electrophilic substitution of 2-tert-butylaniline with I2 on the mild reaction condition. Then, the compatible reaction condition was adopted in the Ullmann coupling reaction of intermediate 2 itself to synthesize diamine 1 due to the existence of sensitive amino groups [33]. That is, Pd(OAc)2 was used directly and reduced in situ into the palladium Pd(0) with catalytic activity by isopropanol among the Ullmann reaction process. Furthermore, the equivalent tetrabutylammonium bromide was added into the reaction mixture as a phase transfer catalyst to conduce to the reaction proceeding homogeneously. Finally the coupling product 1 was obtained through the oxidative addition and reductive elimination reactions. The chemical structure of the resulting diamine 1 was confirmed by 1H NMR and 13C NMR spectra (Fig. 1). The melting point of diamine 1 was observed at 137.0 °C from the DSC analysis curve as a sharp endothermic peak, which is higher than that of MBTBA (82.6 °C).

Synthesis of the rigid diamine 1

1H and 13C NMR spectra of 3,3′-di-tert-butylbenzidine (1)
Polymer synthesis and characterization
The synthetic route of all polyimides is outlined in Scheme 2. In general, polyimides can be prepared from the polycondensation of a diamine and a dianhydride through either a two-step approach or a one-pot method [1]. Our previous research demonstrated that the reactivity of diamine was decreased heavily when the tert-butyl groups were introduced into the ortho-position of amino group in aromatic diamine due to the steric hindrance of large substituent groups [30, 31]. In the same way, the reactivity of diamine 1 was much less than that of benzidine so that a one-pot polycondensation in solution at a high temperature was adopted too in this study.

Synthesis of the polyimides
PMDA was first adopted to react with diamine 1 to synthesize polyimide because PMDA is the cheapest, smallest and most rigid aromatic dianhydride. We anticipated that the resulting polyimide may possess good solubility as well as high T g due to the introduction of pendent tert-butyl groups into the polymer chains reducing the interaction among polyimide backbones as well as the rigid structure of PMDA and diamine 1. Unfortunately, the reaction mixture became non-transparent and some light yellow powder appeared even with a solid content of 1 wt% when the reaction temperature was improved to 100 °C. In other words, the introduction of tert-butyl groups into benzidine is not enough to overcome the rigidity of PMDA and benzidine to produce a soluble polyimide. Then another relatively rigid aromatic dianhydride BPDA was selected to polymerize with diamine 1. When the solid content was maintained lower than 6 wt%, the reactive mixture was kept transparent consistently in the whole polymerization process, i.e., under the following heating program: heated slowly to 85 °C and kept for 12 h, then at 100 °C for 0.5 h, 120 °C for 12 h, 150 °C for 12 h and 220 °C for 12 h. The viscosity of the homogeneous transparent solution increased smoothly during the polycondensation and finally some white fibre-like polyimide was obtained by precipitation in methanol. If the solid content was higher than 6 wt%, some undesired light yellow powder would appear in the polymerization mixture at a time and no high molecular weight polyimide was produced. Other relatively flexible aromatic dianhydrides OPDA, 6FDA and BPADA were able to polymerize with diamine 1 smoothly under the similar procedure and produce some high molecular weight polyimides even when the solid content was larger than 6 wt%. Strangely, it was difficult to produce high molecular weight polyimide when the commonly used aromatic dianhydride BTDA was polymerizaed with diamine 1. In the polymerization process of BTDA and diamine 1, the reaction mixture seemed transparent consistently but its viscosity increased a little. After the polymerization finished and the mixture was cooled to room temperature, the whole polymerization system formed a gel. We conjectured that some chemical cross-linking occurred easily or the liquid - liquid phase separation appeared in the polymerization process [34–38].
The chemical structure of all polyimides was first confirmed by 1H NMR spectra (Fig. 2). The water peak appeared around 3.3 ppm in 1H NMR spectra for all polyimides. It is likely that the deuterated solvent (DMSO-d 6 ) contained a little water or the polyimide films absorbed a little water. The detailed ascription of other signals to the corresponding chemical structure of polyimides is demonstrated in Fig. 2. The FTIR spectra of all polyimides are shown in Fig. 3. All the characteristic peaks for polyimides could be observed at 2,964–2,967 cm−1 (for carbon-hydrogen bond in tert-butyl), 1,779–1,788 and 1,721–1,729 cm−1 (for imide carbonyl), 1,368–1,370 cm−1 (for carbon-nitrogen bond), 1,015–1,104 and 724–752 cm−1 (for imide-ring). The signal of amide carbonyl was not found around 1,650 cm−1, indicating the imidization proceeded completely during the one-pot polycondensation. That is, the byproduct (H2O) was removed rapidly from the reaction system by a nitrogen flow to destroy the equilibrium between water and the imide groups. The small peak around 3,500 cm−1 may be attributed to the polyimide chain end (amino groups) or a little water absorbed by the polyimide film.

1H NMR spectra of the polyimides

FTIR spectra of the polyimides
The number-average molecular weights (M n ) of these polyimides are shown in Table 1. Some high molecular weight aromatic polyimides were still synthesized through a one-pot polycondensation, even though the reactivity of the rigid and hindered aromatic diamine 1 was relatively low. It can be seen from Table 1 that the molecular weights of these polyimides are related to the chemical structure of the aromatic dianhydrides used. The M n of 1-BPADA polyimide was only 42,100 because of the lower reactivity of BPADA, but M n of 1-BPDA reached up to 125,000. The polydispersity indices (PDIs) of all the polyimides were about 2.34 - 3.04.
Thermal properties
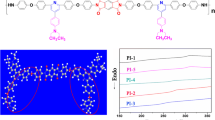
The thermal properties of the polyimides were studied by DSC and TGA under a nitrogen atmosphere. The DSC curves are displayed in Fig. 4 and the corresponding T g s are also shown in Table 1.

DSC curves of the polyimides
As we anticipated, the polyimide prepared from 1-BPDA had a high T g (375.4 °C) due to the rigid structure of biphenyl in diamine and dianhydride. The T g of the polyimide 1-OPDA still reached 350.0 °C even if dianhydride OPDA contained a flexible ether bond. For dianhydride BPADA with two flexible ether bonds, the T g of the resulting polyimide 1-BPADA reached up to 287.2 °C as well, which is considerably higher than that of the commercial polyetherimide (Ultem PEI, T g = 210–215 °C). We inferred that the introduction of pendent tert-butyl group hindered the polyimide chain torsion. The polyimide 1-6FDA also had a relatively high T g of 336.8 °C. Here, the rigid structure of diamine 1 is the key parameter to produce the polyimide with high T g s if the difference of the molecular weights is ignored. The TGA curves of all polyimides are shown in Fig. 5. They were all stable up to 500 °C under the nitrogen atmosphere. In summary, the high T g and excellent heat resistance of the polyimides will not be deteriorated by introducing the pendent tert-butyl group into the backbone.

TGA curves of the polyimides
Solubility
The solubility of the polyimides was determined at 10 mg/mL (polymer/solvent) and room temperature or 60 °C in various organic solvents. The results were summarized in Table 2.
Comparing with the results of our previous research [30, 31], the solubility of the resulting polyimides here decreased somewhat because of the linkage of -CH2- cut off from the diamine with tert-butyl groups. However, the polyimides made from diamine 1 with 6FDA, OPDA, and BPADA still displayed excellent solubility, and even soluble in common organic solvent with low boiling point, such as THF and CHCl3. For the polyimide 1-BPDA with the rigid structure and high T g , it is still soluble in NMP, DMAc and m-cresol at 60 °C. If this polyimide solution is a two-fold reduction in concentration, it could also be soluble in DMAc and m-cresol at room temperature. It can be seen that the incorporation of pendent tert-butyl groups is enough to reduce the interaction of polyimide mainchains and kept the good solubility of these polyimides, even if the rigidity of diamnie is improved. Therefore, these polyimides were processed easily by solution casting to prepare transparent and strong films.
Mechanical behaviors
Polyimide films were obtained by casting the solutions (5 wt % polyimide in NMP) on the clean glass plates and dried under the programmed temperature. The mechanical behavior of these films was measured by a CMT-4104 tensile tester and the results are shown in Table 3. Their tensile strength and tensile modulus ranged from 58.8 to 138.5 MPa and 1.84 to 3.37 GPa, respectively. However, their elongations at break were only from 5.2 to 7.2 %. The relatively low elongations at break indicated that these polyimides were glassy polymers, because this is a typical brittle breakage of glassy polymer at the temperature far lower than their glass transition temperature. Especially for 4b, this film exhibited higher tensile strength (138.5 MPa) and tensile modulus (3.37 GPa) with a elongation at break of 7.2 %, which was attributed to the rigid biphenyl backbone. On the other hand, the lower tensile strength of 4f was ascribed to its relatively low molecular weight (M n = 4.21 × 104).
X-ray diffraction data
The X-ray diffraction curves of the polyimide films were displayed in Supporting Information Fig. S1. Only one wide diffuse X-ray peak could be observed in each curve and further confirmed all of these polyimides are amorphous glassy polymers due to the incorporation of bulky tert-butyl decreasing the well ordered chain packing. In addition, the amorphous state also improved the solubility of polyimides.
UV–vis spectra
The UV–vis spectra of the polyimide films are shown in Fig. 6. The polyimide films were prepared from solution casting on quartz pieces at a concentration of 0.5 wt % in NMP and showed a high transmission above 90 % in a wavelength range of 400–700 nm. This can be ascribed to the tert-butyl moiety increased the intermolecular distance and decreased the interactions among polyimide chains to result in good optical transparency. Especially for the film of polyimide 4e (1-6FDA), it has the most excellent optical transparency because of the lower polarization ability of the C-F bond weaken the chain-to-chain cohesive force. On the other hand, it seemed that the transparency of these films increased with the flexibility of the corresponding chemical structure of polyimides improved.

UV–vis spectra of the polyimide films
Conclusions
Some soluble polyimides with high T g s were synthesized from the aromatic diamine 1 and various aromatic dianhydrides. Our results further confirm that the aromatic diamine containing two tert-butyl groups has lower reactivity, but soluble polyimides with high molecular weights are still produced by a one-pot polycondensation. The incorporation of pendent tert-butyl groups is enough to reduce the interaction of polyimide mainchains and keep the good solubility of these polyimides, even if the rigidity of the diamnie is improved. They can be processed easily by solution casting to prepare transparent, flexible, and tough films. These soluble polyimides with high T g s are potential candidates for high performance polymer materials.
Abbreviations
- BPDA :
-
3,3′,4,4′-biphenyltetracarboxylic dianhydride
- OPDA :
-
3,3′,4,4′-diphenylether tetracarboxylic dianhydride
- 6FDA :
-
2,2-bis(3,4-dicarboxyphenyl)-1,1,1,3,3,3-hexafluoropropane dianhydride
- BPADA :
-
2,2-bis [4-(3,4-dicarboxyphenoxy)phenyl]propane dianhydride
References
Ghosh MK, Mittal KL (1996) Polyimides: fundamentals and applications. Marcel Dekker, New York
Kennedy BW (1972) U.S. Patent 3 700 538
Katz M, Theis RJ (1997) IEEE Electr Insul Mag 13:24–30
Irwin RS, Sweeny W (1967) J Polym Sci Part C Polym Symp 19:41–48
Makino H, Kusuki Y, Harada T, Shimazaki H, Isida T (1985) U.S. Patent 4 528 004
St Clair TL, Progar BJ (1979) In the enigma of the eighties: environment, economics, energy: 24th national SAMPE symposium and exhibition. The society, San Francisco
Liaw DJ, Wang KL, Huang YC, Lee KR, Lai JY, Ha CS (2012) Prog Polym Sci 37:907–974
Baise AI (1986) J Appl Polym Sci 32:4043–4048
Feld WA, Ramalingam B, Harris FW (1983) J Polym Sci A Polym Chem 21:319–328
Hsiao SH, Chang YM, Chen HW, Liou GS (2006) J Polym Sci Part A Polym Chem 44:4579–4592
Zhang QY, Li SH, Li WM, Zhang SB (2007) Polymer 48:6246–6253
Hsiao SH, Wang HM, Chou JS, GuoW, Lee TM, Leu CM, Su CW (2012) J Polym Res 19:9757
Thiruvasagam P, Vijayan M (2012) J Polym Res 19:9845
Thiruvasagam P (2012) J Polym Res 19:9965
Hsiao SH, Wang HM, Chang PC, Kung YR, Lee TM (2013) J Polym Res 20:154
Hsiao SH, GuoW J, Tsai TH, Chiu YT (2014) J Polym Res 21:391
Kumar SV, Yu HC, Choi J, Kudo K, Jang YH, Chuang CM (2011) J Polym Res 18:1111–1117
Hsiao SH, Huang TL (2004) J Polym Res 11:9–21
Tanaka K, Kita H, Okano M, Okamoto KI (1992) Polymer 33:585–592
Chung CL, Tzu TW, Hsiao SH (2006) J Polym Res 13:495–506
Yang CP, Chen YC, Hsiao SH, Guo W, Wang HM (2010) J Polym Res 17:779–788
Rusanov AL, Shifrina ZB (1993) High Perform Polym 5:107–121
Harris FW, Sakaguchi Y, Shibata M, Cheng SZD (1997) High Perform Polym 9:251–261
De Abajo J, De La Campa JG (1999) Adv Polym Sci 140:23–59
Liaw DJ, Liaw BY (1996) Polym J 28:970–975
Liaw DJ, Liaw BY (1998) J Polym Sci A Polym Chem 36:2301–2307
Yang CP, Hsiao SH, Yang HW (1998) Polym J 30:723–729
Yang CP, Hsiao SH, Yang HW (1999) Macromol Chem Phys 200:1528–1534
Havva Y, Lon JM (1998) Polymer 39:3779–3786
Huang W, Yan DY, Lu QH (2001) Macromol Rapid Commun 22:1481–1484
Huang W, Yan DY, Lu QH, Tao P (2002) J Polym Sci A Polym Chem 40:229–234
Zhang QM, Liu HM, Li CL, Fang XJ, Liu H, Tong XF (2009) CN Patent 101 531 598
Hassan J, Penalva V, Lavenot L, Gozzi C, Lemaire M (1998) Tetrahedron 54:13793–13804
Lin AA, Sastri VR, Tesoro G, Reiser A, Eachus R (1988) Macromolecules 21:1165–1169
Vanherck K, Koeckelberghs G, Vankelecom IFJ (2013) Prog Polym Sci 38:874–896
Cheng SZD, Lee SK, Barley JS, Hsu SLC, Harris FW (1991) Macromolecules 24:1883–1889
Lee SK, Cheng SZD, Wu Z, Lee CJ, Harris FW, Kyu T, Yang JC (1993) Polym Int 30:115–122
Harris FW, Hsu SLC (1989) High Perform Polym 1:3–16
Acknowledgments
The authors gratefully acknowledge the financial supports provided by the National Basic Research Program (No. 2014CB643604, 2013CB834506, 2012CB821500, 2009CB930400), National Natural Science Foundation of China (No. 21174086, 21074069, 91127047).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 87 kb)
Rights and permissions
About this article

Cite this article
Yi, L., Li, C., Huang, W. et al. Soluble aromatic polyimides with high glass transition temperature from benzidine containing tert-butyl groups. J Polym Res 21, 572 (2014). https://doi.org/10.1007/s10965-014-0572-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-014-0572-7