
Abstract
The first ionization constants of phosphoric acid and acetic acid have been measured in H2O and D2O from T = 373 K to T = 573 K and p = 11.5 and 20 MPa to yield accurate values of the deuterium isotope effect. Sequential conductivity measurements using a unique high-precision flow-through AC conductance instrument were made on dilute (m ≤ 10–2 mol·kg−1) aqueous solutions of phosphoric acid, acetic acid, potassium dihydrogenphosphate, sodium acetate, potassium hydroxide, sodium hydroxide, hydrochloric acid, potassium chloride and sodium chloride in light and heavy water under the same experimental conditions (temperature, pressure, flow-rate), so that systematic experimental errors between the two solvents would cancel. The experimental molar conductivities of potassium dihydrogenphosphate, sodium acetate, hydrochloric acid, and the corresponding chloride salts were used to calculate the molar conductivities for the fully dissociated acids [λ(D+), λ(D2PO −4 ) and λ(CH3COO−)]. Together with the molar conductivities measured for partially ionized acids, Λ(D3PO4) and Λ(CH3COOD), these yielded values for the degree of dissociation, α, and the ionization constants, pKa1. The iterative process was repeated at each temperature in both H2O and D2O where the Fuoss-Hsia-Fernández-Prini (“FHFP”) and the Quint-Viallard (“QV”) equations were used to correct for ionic strength. The resulting values of pKa1 for phosphoric acid in H2O agree with those reported from conductivity studies by previous works over the entire temperature range and with low temperature potentiometric studies to within the combined experimental uncertainties. The results for pKa1 above 298.15 K in D2O are the first to be reported in the literature. The new values for pKa(CH3COOD) yield more accurate values for the deuterium isotope effect on the ionization constant of acetic acid than those reported in our previous work (Erickson et al. in J. Phys. Chem. B. 123:9503–9506, 2019). The single-ion limiting conductivities for dihydrogenphosphate and acetate in D2O, λ(D2PO −4 ) and λ(CH3COO−), were found to be the same as those in H2O once corrected for viscosity effects, confirming previous observations for other ions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
An accurate understanding of the pH and chemical speciation of aqueous systems at high temperature is critical to optimise the plant chemistry regime of nuclear power plants and establish safety margins. The Canadian CANDU-PHWR (“CANada Deuterium Uranium Pressurized Heavy Water Reactor”) design is unique compared to other Pressurized Water Reactors (PWRs) in that it uses D2O rather than H2O in the primary coolant system as a neutron moderator and heat transport medium [1]. To optimize PHWR chemistry regimes and safety margins, experimental values for the equilibrium constants of acid ionization and metal oxide hydrolysis reactions in heavy water at high temperature and pressure are required. A few high temperature aqueous systems have been reported [2,3,4] with the data being much more limited for D2O systems. Understanding the deuterium isotope effects on equilibrium constants for a variety of chemical systems over an extended temperature range will help to probe differences in the solvation behaviour of light and heavy water to develop accurate models of their reactions under CANDU primary coolant conditions.
A great deal of progress has been made towards understanding these effects at ambient temperatures [5,6,7,8,9,10,11]. The increase in the molar mass of heavy water causes small but detectable differences in the properties of the solvent, and as temperature increases the average number and strength of the hydrogen bonds in both H2O and D2O weaken, resulting in drastic changes in the physical properties of both solvents. Experimental deuterium isotope effects on ionization reactions, ΔpK = pKa,D2O−pKa,H2O, have been reported at ambient conditions for many organic and inorganic species [5,6,7,8,9,10,11,12], where ΔpK values ranged between 0.2 and 0.7. At higher temperatures accurate measurements remain scarce, with only a few studies of acid–base ionization having been reported above 373 K [13,14,15,16,17,18,19,20,21,22].
This work adds to previous investigations carried out at the University of Guelph [16,17,18,19,20,21,22], aimed at measuring the small differences in equilibrium constants between light and heavy water over an extended range of temperatures and pressures with sufficient accuracy that the deuterium isotope effects can be modeled. This study reports measurements of the phosphoric acid first ionization constant in H2O, Ka1,H2O, and D2O, Ka1,D2O, from 298 to 571 K at 20 MPa using a unique custom-made high-temperature flow AC conductivity instrument [23, 24], where the experiments were designed to measure the difference in molar conductivity between solutions in D2O and H2O directly, so that most systematic errors would cancel. The measurements also yielded values for the single-ion limiting molar conductivities, λ°, of K+, Cl−, Na+, D2PO −4 /H2PO −4 , CH3COO−, D3O+/H3O+ and OD−/OH− in D2O and H2O at temperatures up to T = 573 K. The additional measurements on the ionization constant of acetic acid were made to reduce the uncertainties on its deuterium isotope effect reported in our previous study [18, 19]. The conductivity data were analyzed with the Fuoss-Hsia-Fernández-Prini (“FHFP”) model [25] and the Quint-Viallard (“QV”) [26,27,28] equations following Conrad et al. [29].
2 Experimental
2.1 Chemicals and Solution Preparation
Aqueous stock solutions of 1.1905 mol·kg−1 H3PO4 in H2O and 0.8679 mol·kg−1 D3PO4 in D2O were prepared from purchased phosphoric acid aqueous solutions (Fisher Scientific, #A242P-500, 85 wt %) and (Sigma Aldrich, #176,753, 85 wt % in D2O, 98 atom % D), respectively. Both solutions were standardized against a standard NaOH solution at 298 K to ± 0.1%. Aqueous stock solutions of 0.1299 mol·kg−1 HCl, 0.4677 mol·kg−1 DCl, 0.2433 mol·kg−1 KOH, and 0.1355 mol·kg−1 KOD were prepared by dilution from purchased solutions (HCl, Sigma Aldrich, #318965, 0.1 N standard; DCl, Sigma Aldrich, #543047, 35 wt % in D2O, 99 atom % D; KOH, Fisher Scientific, #SS254-4, 50 wt %; KOD, Sigma Aldrich, #176761, 40 wt % in D2O, 98 + atom % D). The HCl and DCl solutions were standardized against a standard NaOH solution, and the KOH and KOD solutions were standardized against potassium hydrogen phthalate (KHP, C8H5O4K, Fisher Scientific, #AC17712, 99.99%) to ± 0.1% at 298 K. Stock solutions of 0.1163 mol·kg−1 KCl (Fisher Scientific, #BP366-500, > 99%) in H2O and 0.1134 mol·kg−1 KCl in D2O, 0.1011 mol·kg−1 KH2PO4 (Sigma Aldrich, #P0662, > 99%) in H2O, and 0.09297 mol·kg−1 KD2PO4 (Sigma Aldrich, #329916, 98 atom % D) in D2O were prepared by mass from their salts using a balance accurate to ± 0.00002 g with buoyancy corrections and accuracy better than ± 0.2%. KHP, KCl, and KH2PO4 were dried at 403 K, 573 K, and 393 K respectively until a constant mass was achieved.
Degassed and deionized water from a Millipore Direct-Q 5 water purification system (resistivity 18.2 MΩ·cm) was used to prepare the H2O solutions. The heavy water used in the D2O solution preparation was donated from Ontario Power Generation Inc. and determined to be > 99.8 mol% D with 1H NMR, using a standardized D2O solution of acetic acid (CH3COOD, Sigma Aldrich, #537020, 99.85%) as the internal proton reference. The solutions in D2O were prepared in a glove bag purged with argon, which was continually kept under positive argon pressure. Stock solutions were diluted to a concentration of ~ 10–3 mol·kg−1 by mass, to a relative precision of ± 0.01%, and stored in sealed Pyrex glass or HDPE Nalgene bottles under a positive argon atmosphere to prevent contamination from atmospheric CO2(g).
2.2 Impedance Measurements and Experimental Design
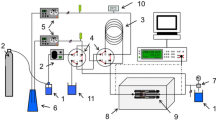
The experimental design was based on our recent acetic acid study [18, 19]; here, solutions of phosphoric acid, acetic acid, potassium dihydrogenphosphate, sodium acetate, hydrochloric acid and potassium or sodium chloride, in both light and heavy water, were injected in sequence at each temperature, pressure, and flow rate, so that systematic errors in the experimental molar conductivities of solutions in the two solvents would cancel, thus yielding more precise values of the deuterium isotope effect, ΔpK = pKa1,D2O—pKa1,H2O. Experiments were performed for dilute aqueous solutions of H3PO4/D3PO4, CH3COOH/CH3COOD, HCl/DCl, KOH/KOD, KCl, NaCl, NaCH3COO, and KH2PO4/KD2PO4 in both H2O and D2O (~ 10–4 to ~ 10–3 mol·kg−1) from T = 373 K to 573 K at a constant pressure p ~ 11.5 or 20 MPa with a high-temperature, high-pressure AC conductivity flow of cell used in our earlier studies [24, 30,31,32,33,34]. The temperature of the cell was controlled to ± 0.15 K over the several days required for conductivity measurement and recorded to ± 0.02 K. High-performance liquid chromatographic (HPLC) pumps were used to inject solutions into the flow conductivity cell, at a rate of 0.5 cm3·min−1. The pressure was controlled by a back-pressure regulator (Circle Seal Controls Inc. model BPR21U25512 6000 psi) located at the end of the flow line and measured to an accuracy of ± 0.01 MPa. A set of Pyrex glass bottles or high density polyethylene Nalgene bottles (for KOH, KOD, NaOH and NaOD), equipped with KIMAX GL-45 gastight tops, contained the test solutions in both light and heavy water, and the two solvents, H2O and D2O, for injection into the high-pressure flow AC conductivity instrument. The solutions and solvents in the flasks used for the feed system were kept under a positive argon pressure for the duration of the experimental runs.
Complex impedances, Z(ω) = ZRe(ω)−j·ZIm(ω), of the aqueous solutions were measured as a function of concentration and the angular frequency (ω) at each temperature. Impedance spectra were collected at nine frequencies in the range 100 to 10 kHz, to yield values for both real ZRe(ω) and imaginary ZIm(ω) components. AC impedance data (at 100, 200, 500, 1000, 2000, 4000, 6000, 8000 and 10 000 Hz) were collected for the following sequence of solutions for the phosphoric acid measurements: H2O, D2O, NaCl/H2O, NaCl/D2O, KCl/H2O, KCl/D2O, H3PO4, D3PO4, KH2PO4, KD2PO4, HCl, DCl, KOH, KOD, NaCl/H2O, NaCl/D2O, D2O and H2O, and similarly, for the acetic acid measurements: H2O, D2O, NaCl/H2O, NaCl/D2O, CH3COOH, CH3COOD, CH3COONa/H2O, CH3COONa/D2O, HCl, DCl, NaOH, NaOD, NaCl/H2O, NaCl/D2O, D2O and H2O. Eighty to one hundred impedance measurements were taken with a computer over a time span of 50 min or more. Each sample injection was followed by a large injection of de-ionized water from the main reservoir, typically 80 mL, to rinse the equipment until the cell conductance had returned to its baseline value. The resistance of the solution was determined from the high frequency limit of ZRe(ω) using Eq. 1:
where Rs is the solution resistance that we seek to measure, and b1 and n are fitting parameters. Details on the extrapolation method and the experimental uncertainty limits in ZRe(ω) and ZIm(ω) are discussed in Ref. [35].
2.3 Experimental Conductivities and Solvent Correction
Experimental solution conductivities (κsolnexp= Rs / kcell) were determined from the resistances of the aqueous solution (Rs) after a calibration of the conductivity cell. The procedures used for calibrating the instrument and determining the cell constant (kcell) have been reported in previous studies [18, 19, 24, 34, 36]. Temperature-dependent cell constants used in this work are reported in Tables 1 and 2. To check the accuracy of the calibration, two NaCl solutions (in H2O and in D2O) were run as internal standards before and after the series of other electrolytes at each temperature. As shown in Tables 3, 4 and 5 the values agreed with one another to within ± 1 % or better, which is considered to be the precision of our conductivity measurements. Limiting conductivity values, Λ°(NaCl), were derived from these measurements, and were found to agree with literature results [24] to within less than ± 3% which is considered to be the experimental uncertainty of our conductivity measurements. Results are shown in Figure 1.
As in our previous studies, the data treatment to extrapolate experimental conductivities to infinite dilution is based on the theoretical Fuoss–Hsia–Fernández-Prini (“FHFP”) conductivity model [25]. In this work the Quint-Viallard (“QV”) conductivity model [26,27,28] was also used to confirm the results from Conrad et al. [29]. The full details for the QV treatment are reported in Ref. [29]. Experimental solution conductivities, κ expsoln , were corrected for impurities within the solvent to yield the true solution conductivities, κ. The solvent correction is different if the considered electrolyte is an acid, a base, or a salt. For simple salts, experimental solution conductivities were corrected for impurities within the solvent using the expression:
where solution conductivities of acids and bases, κ expsoln , were corrected following:
because these chemicals shift the auto-ionization equilibrium state of the solvent, and thus affect its contribution to the solution conductivity. In Eqs. 2 and 3, κ expw is the experimental conductivity of water and κ purew is the conductivity of pure water calculated assuming the molar conductivity of H+ (H3O+) and OH− equal their limiting values. Temperature dependent values of λ°(H3O+) and λ°(OH−) were calculated using the Marshall reduced density relationship [37], and for Kw,H2O they were obtained from the equation of state (EOS) reported by Marshall and Franck [38] (using EOS from Refs [33] or [39] lead to insignificant differences in the final results). D2O values of Λ °w [Λ °w = λ°(D3O+) + λ°(OD−)] were calculated using the Marshall reduced density relationship [37] and applying a correction for viscosity effects [34] using Walden’s rule. Values of Kw,D2O were calculated using a density model fit to the experimental data from Mesmer and Herting [15]:
where T is the temperature in Kelvin and ρD2O the solvent density in g·cm−3. Values for ρD2O were taken from [40].
Finally, molar conductivities for each solution, Λexp, were calculated from the corrected conductivities, κ, using the relationship:
where κ is in SI units of S·m−1 and concentration, c, is in mol·dm−3. In this study, these were converted to units of S·cm−1 and mol·cm.−3, respectively. Solution molarities were converted into molarity values following the methodology previously reported by Erickson et al. [18, 19]
Molalities, molarities, conductivities κ, κ expw and κ purew , and the corresponding experimental molar conductivities, Λexp, for each aqueous solute are tabulated in Tables 1 and 2, along with the average temperature (IPTS-90) and pressure. The uncertainties in these tables were estimated using procedures identical to those reported in Refs [18, 19, 29].
3 Conductivity Data Treatment and Methodology to Calculate ΔpK
3.1 Conductivity Data
3.1.1 Limiting Molar Conductivities
The limiting conductivity data for each aqueous electrolyte, Λ°, in both light and heavy water were calculated using both the FHFP [25] and QV [26,27,28] models for fully dissociated electrolytes. Bianchi et al. [41] have evaluated the different theoretical equations [25, 42,43,44,45] for the concentration dependence of molar conductivity, Λ, in H2O and have recommended the FHFP treatment for 1:1 electrolytes which yields values to within an accuracy of ~ 1% over the range 0–0.1 mol·cm−3. Although the QV model does not reduce to the FHFP model for 1:1 electrolytes [29], the results presented below indicate that the QV model gives the same level of accuracy for dilute electrolytes between 298 and 573 K, with the added advantage that it can be used for non-symmetrical electrolytes in H2O and D2O.
As in our previous work [18, 19], experimental molar conductivity values in light and heavy water were calculated from Eq. 5 and values for the molarities, c, in H2O and D2O were calculated from the molalities, m, and solution density. Solution densities were calculated from the density of pure solvents, ρH2O and ρD2O, and the HKF standard partial molar volumes, V°, [46] in light water (the uncertainties associated with approximating the apparent molar volume with V° calculated from the HKF model are less than the experimental uncertainties). For the phosphoric acid aqueous solutions, at each temperature, values of V° [V° = α·V°(H+) + α·V°(H2PO4−)—(1−α)·V°(H3PO4)] were calculated through an iterative process where initial estimates for the degree of dissociation, α, in Eq. 15 were taken from Mesmer and Baes’ results [47] for H3PO4 (aq) assuming a temperature independent value of ΔpKa1 = 0.21 [12] for D3PO4(aq). For CH3COOH (aq) and CH3COOD (aq), the initial estimates for the degree of dissociation were calculated using Mesmer et al. [48] for pKa and Erickson et al. [18, 19] for ΔpKa.
Numerical values for the limiting molar conductivities, Λ°, for each electrolyte measured in this work are reported in Tables 1 and 2.
3.1.2 Single-Ion Limiting Molar Conductivities
Values for the limiting molar conductivities of the chloride single ion, λ°(Cl−), in both light and heavy water were determined from the molar conductivity data listed in Tables 1 and 2, using KCl and NaCl transference numbers calculated following the methods described by Plumridge et al. [34]:
where T is the temperature in K.
The limiting conductivity for the chloride anion was then determined from:
using the Eqs. 6–9 and the experimental conductivities of aqueous KCl or NaCl to yield single ion limiting conductivities for K+ and Na+ at each experimental condition. The other single ion limiting conductivity values were deduced from the KCl or NaCl molar conductivities using Kohlrausch’s law. Single ion limiting conductivity data for K+, Na+, Cl−, CH3COO−, H2PO −4 /D2PO −4 , H3O+/D3O+ and OH−/OD− in light and heavy water are tabulated in Tables 6 and 7 together with the values of t°(Cl−) used to split the molar conductivity results.
3.2 Equations Used to Calculate ΔpK
3.2.1 Equations Used to Calculate Phosphoric Acid and Acetic Acid First Ionization Constants in H2O and D2O
Phosphoric acid can undergo three successive dissociation reactions in water according to the following equilibria:
where α = α1 + α2 + α3 is the total degree of dissociation, and α1, α2, α3 are the fractions of dissociation for Eqs. 11–13.
The equilibrium constant for the first ionization of phosphoric acid (expressed in the molarity basis), \({K}_{{\mathrm{a}}_{1}\text{,}\mathrm{c}}\), which is the equilibrium constant we seek to measure, can be calculated from the fractions of dissociation, the stoichiometric concentration of phosphoric acid, c, and the activity coefficients:
where \({\gamma }_{{{\text{H}}_{2}{\text{PO}}}_{4}^{-}}\), \({\gamma }_{{\text{H}}^{+}}\) and \({\gamma }_{{{\text{H}}_{3}{\text{PO}}}_{4}}\) are the activity coefficients of the H2PO −4 , H+ and H3PO4. Optimal experimental conditions where the contributions from reactions 12 and 13 are negligible compared to reaction 11 (i.e. α ≈ α1, and α2 ≈ α3 ≈ 0) were determined by estimating the chemical speciation between 373 and 573 K using equilibrium constants for Eqs. 11–13 taken from the computer program SUPCRT [46] and neglecting activity coefficient effects (i.e. γi = 1).
Under these optimized experimental conditions, the equilibrium constant for the first ionization reaction of phosphoric acid, \({K}_{{\mathrm{a}}_{1}\text{,}{\mathrm{H}}_{2}{\text{O}}}\), can be calculated assuming:
where the degree of dissociation can be derived from the conductivity results:
In Eq. 15, γ± is the mean activity coefficient of the electrolyte solution based on the hypothetical 1 mol·L−1 standard state extrapolated to infinite dilution; values of γ± were calculated using the extended Debye–Hückel equation [25, 34]. In Eq. 16, Λexp is the experimental molar conductivity of phosphoric acid and [λ(H+) + λ(H2PO −4 )] corresponds to the molar conductivity of the fully dissociated acid (at the same ionic strength I = α·c).
Values of [λ(H+) + λ(H2PO −4 )] were calculated from the experimental molar conductivity values of hydrochloric acid, potassium dihydrogenphosphate and potassium chloride, according to Eq. 17,
where the experimental molar conductivities were corrected for small differences in ionic strength using the Fuoss-Hsia-Fernández-Prini (“FHFP”) [34] and the Quint-Viallard (“QV”) conductivity equations [26,27,28] for fully dissociated electrolytes assuming Bjerrum’s definition for the distance of closest approach.
The exact same methodology was adopted to determine the equilibrium constant for the first ionization of phosphoric acid in D2O. In the analysis, the properties of water (density, ρw, viscosity, ηw and static dielectric constant, εw) were calculated from the equations of state recommended by the International Association for The Properties of Water and Steam (IAPWS) using software distributed by the National Institute of Standards and Technology (NIST) [40]. The software does not include an equation of state for the dielectric constant of D2O, and corresponding values were calculated following the approach developed by Trevani et al. [16]. Briefly, the method is based on the assumption that values of εw in H2O and D2O at each temperature should be equal at the same number density of water molecules, as suggested from the high temperature study by Okada et al. [49]; values for εw in H2O were calculated from the Uematsu-Franck equation [50].
A similar approach was taken to examine the acetic acid data to yield temperature-dependent ionization constants for:
with:
3.2.2 Hydrolysis Corrections to KH2PO4 and NaCH3COO Conductivity Data
The present method is based on measurements made at a single ionic strength for each electrolyte and can only be applied at conditions where ion association is negligible. In this study, experimental molar conductivities were measured at an ionic strength of ~ 10–3 mol·kg−1. The determination of λ(K+) + λ(H2PO −4 ) from Λexp(KH2PO4) and of λ(Na+) + λ(CH3COO−) from Λexp(NaCH3COO) was complicated by the hydrolysis of H2PO4−/CH3COO−, according to the reactions:
To address this complication, KH2PO4 experimental molar conductivities were corrected at each temperature following the method described by Tsurko et al. [51], using the following equation:
where α’ is the primary degree of dissociation and β is the fraction of undissociated molecules of phosphoric acid as defined by Tsurko et al. [51]. The molality of each species at chemical equilibrium was calculated using the pKa1 and ΔpKa1 values determined in this work together with the equilibrium constants reported by Mesmer et al. [47] for the second ionization of phosphoric acid, and ΔpKa2 = 0.468, assumed to be temperature independent [12]. In these calculations, the molar conductivity for HPO 2−4 , DPO 2−4 , H+, D+, K+/H2O and K+/D2O were calculated using the Debye–Onsager limiting law [24, 52], using limiting conductivities λ° from Conrad et al. [29] for K+/H2O and K+/D2O and estimates derived from the data of Muccitelli and DiAngelo [53] for HPO 2−4 . The values for λ°(DPO 2−4 ) were estimated from those of λ°(HPO 2−4 ) by correcting the viscosity effect difference between both solvents based on insights from Ref. [34]. The hydrolysis corrections for sodium acetate were done following the same methodology. In all cases, corrections were < 2.5% for acetate and < 0.5% for hydrogen phosphate. (Table 8)
3.2.3 Ionization Constants in H2O and D2O
Tables 8 and 9 report temperature-dependent ionization constants for phosphoric acid and acetic acid in H2O and D2O calculated from the conductivity results following the methodology described above. The equilibrium constants are expressed in terms of the hypothetical 1 molar (mol·L−1 of solution) standard state (pKa,c = −log10 Ka,c), and in terms of the hypothetical 1 molal (mol·kg−1 of water) standard states (pKa,m = −log10Ka,m), where:
here Δνi is the difference in stoichiometry between the aqueous product and reactant species. For example, for Eq. 11 Δνi = +1, and Ka,m = Ka,c/ρw.
Experimental values for the difference in pK, ΔpK = \(\text{p}{K}_{{\mathrm{a1},}{\text{D}_{2}\text{O}}}-\text{p}{K}_{{\mathrm{a1},}{\text{H}_{2}\text{O}}}\), are also listed in Tables 9 and 10. Standard uncertainties were estimated using procedures identical to those reported in Ref. [29]. Hydrolysis corrections were calculated by estimating the fractions of dissociation using the methodology described in Sect. 3.1, where the temperature-dependent values for limiting conductivities of OH− and OD− were taken from Refs [29] and [54] (see discussion below in Sect. 4.1).
4 Results and Discussion
4.1 Limiting Conductivity Results
4.1.1 Molar Limiting Conductivity
To illustrate the accuracy of the present results, experimental molar conductivities for hydrochloric acid, potassium chloride and potassium hydroxide in H2O and D2O are compared in Fig. 2 to other literature studies [18, 19, 54,55,56] which used the same experimental technique. The limiting molar conductivities of HCl, DCl, KCl/H2O, KCl/D2O and KOH agree with other literature studies [18, 19, 54,55,56] to within the combined experimental uncertainties between 373 and 573 K. The KOD results measured at p = 20 MPa differ from those reported by Arcis et al. [54]; the viscosity effect on the Λ°(KOD) data measured in this work is not smooth, suggesting an experimental problem with the solution preparation, as illustrated by the discrepancy for the two data points measured at 373 K and 423 K. These data were originally measured for the hydrolysis correction and were discarded. The viscosity effect on the limiting molar conductivity of KH2PO4 and KD2PO4 at 20 MPa was also found to be irregular and supplementary measurements were conducted at 11.5 MPa to replace these data (the 20 MPa KH2PO4/KD2PO4 data were not considered in the final analysis because of problems with the solution preparation, confirmed by the subsequent set of experiments at 11.5 MPa). No literature comparisons were done as no experimental limiting conductivity data have been reported for KH2PO4 in H2O or D2O above 323 K. Except for a few outliers, the NaCH3COO, NaCl (in H2O and D2O), NaOH and NaOD results were found to be consistent with Refs, [34, 54] and [57] to within the experimental uncertainties.

a Experimental limiting conductivity of NaCl from T = 298 K to T = 598 K at p = 20 MPa: filled circle, this work; solid line, best fit from Zimmerman et al. [84]; b Experimental limiting conductivity of NaCl from T = 298 K to T = 573 K at p = 11.5 MPa: open circle, this work (run 1); open diamond, this work (run 2); solid line, best fit from Zimmerman et al. [84]

Limiting molar conductivity of a hydrochloric acid, b potassium chloride, c potassium hydroxide in H2O and D2O from 298 to 573 K:open inverted triangle, this work; filled inverted triangle, this work; open circle, Ref. [5]; filled circle, Ref. [5]; open diamond, Refs [31, 32] ; open square, Refs. [21, 22]; filled square, Refs [21, 22] solid line, fit [5] (open and filled symbols refer to H2O and D2O data, respectively)
4.1.2 Single Ion Limiting Conductivity
Single ion limiting conductivities are compared with other literature results for dihydrogenphosphate [51] and acetate [18, 19, 57,58,59,60] in Fig. 3.

Limiting conductivity of a dihydrogenphosphate and b acetate in H2O and D2O versus solvent viscosity from 288 to 573 K: a open circle, this work (H2O); open diamond, [75]; filled circle, this work (D2O); solid line, Eq. 25 (psat); b open diamond, [36], open inverted triangle, (H2O) [11]; filled inverted triangle, (D2O) [11]; multiplication sign , [81]; open square, [48]; red open triangle, (H2O) [21, 22] (not included in the fit); red filled triangle (D2O) [21, 22], (not included in the fit); open circle, this work (H2O); filled circle, this work (D2O); solid line, Eq. 25 (psat)
The new acetate results compare much better with previous literature studies [57,58,59,60] in H2O and D2O (Fig. 3) over the whole temperature range, and confirmed the previous measurements by Erickson et al. [18, 19] were not accurate.
The 11.5 MPa H2PO −4 conductivity data in light water are consistent with the low temperature data from Tsurko et al. [51], however it became evident that there was an issue with the H2PO −4 and D2PO −4 conductivity data measured at 20 MPa. No other data could be identified for comparison; because of the limited concentration range investigated and the lower accuracy of the static method the KH2PO4 and NaH2PO4 conductivity measurements from Ref. [53] could not be used to derive accurate limiting conductivity data for H2PO −4 . The H2PO −4 and D2PO −4 conductivity measurements at 20 MPa were therefore discarded. The discrepancy at 20 MPa is believed to be due to errors in the solution preparation combined with non-optimal experimental concentrations. Recognising that it was not practical to duplicate the full set of phosphoric acid measurements, the analysis that follows used the H2PO4− and D2PO −4 limiting conductivities measured at 11.5 MPa and corrected to 20 MPa using fitted values (Eq. 25). Because no measurements were made at 298 K and 11.5 MPa it was decided to use Tsurko’s 298 K data in the fit that is presented below; Tsurko’s 298 K data was also used for D2PO −4 once corrected for viscosity effects [34]. This approach is supported by our recent results [29, 34, 54].
The semi-empirical model proposed by Plumridge et al. [34] (Eq. 25) was fitted to the experimental limiting conductivities for H2PO −4 , D2PO −4 and CH3COO− (in H2O and D2O) reported in Tables 6 and 7.
where ρw is the solvent density in kg·m−3 and ηw is the solvent viscosity in Poise (P). Fitting parameters listed in Table 10, some of which had been reported in our earlier studies [29, 34, 54], were found to represent the temperature dependence of the limiting conductivity in Tables 6 and 7 to within the experimental uncertainties, and no attempts were made to further fine-tune the parameters.
4.2 Equations of State for the First Ionization Constant of Phosphoric Acid and the Ionization Constant of Acetic Acid in H2O
Following [20, 21, 29] the equilibrium constants in light water for the ionization of H3PO4 and CH3COOH were fitted as a function of temperature and molar volume by using a modified form of the ‘density’ model reported by Mesmer et al. [61]:
where p1, p2, and p3 are adjustable fit parameters, T is the temperature in Kelvin, and the solvent density ρH2O has been replaced by its molar volume, \({V}_{{\mathrm{H}}_{2}\mathrm{O}}^{*}= {\mathrm{M}}_{{\mathrm{H}}_{2}\mathrm{O}}/{\rho }_{{\mathrm{H}}_{2}\mathrm{O}}\), in cm3·mol−1. The thermodynamic justification for using molar volumes of the two solvents rather than densities in this model is discussed in more detail in Ref. [20].
Equation 26 was fitted to the experimental values of pKa,m tabulated in Tables 9 and 10 for H3PO4 and CH3COOH along with literature sources listed in Tables 11 and 12. Parameters are tabulated in Tables 11 and 12. Our experimental pKa,m values are compared with other literature studies in Figs. 4 and 5 for H3PO4 [62,63,64,65,66,67,68] and for CH3COOH [18, 19, 48, 57, 58, 69,70,71,72,73,74,75,76,77,78].

a Phosphoric acid first ionization constant in H2O, pKa1,m vs T from 288 to 573 K, b deviation plot between experimental and fitted values (pKexp–pKfi): Literature data (p = psat) open square, [49]; filled diamond, [56]; open hexagon, [47]; open circle, [70]; open triangle, [60]; open inverted triangle, [67]; filled square, [33]; red open diamond, [69]; open star, [71] (not included in the fit); filled circle, This Work (p = 20 MPa); solid line, Eq. 26 (p = 20 MPa); doted solid line, Eq. 26 (p = psat)

a Acetic acid ionization constant in H2O, pKa,m vs T from 288 to 623 K, b deviation plot between experimental and fitted values: plus, [43]; open inverted triangle, [57] recalculated by [81]; multiplication sign , [29]; open star, [71]; open diamond, [35]; filled diamond, [26]; filled inverted triangle, [20]; open circle, [25]; filled triangle, [24]; filled hexagon, [59]; open square, [52]; open hexagon, [81]; filled square, [73]; open triangle, [21, 22]; filled circle, This Work; solid line, Eq. 26 (p = 20 MPa); doted solid line, Eq. 26 (p = psat)
4.2.1 Phosphoric Acid pK a,m in H2O
There have been several studies of the first ionization constant of phosphoric acid in H2O [47, 62,63,64,65,66,67,68, 74]. Briefly, Nims [62], Schwarzenbach et al. [74], Glasoe and Long [75], McDougall and Long [63], Salomaa et al. [64], Paabo and Bates [65] and Mesmer and Baes [47] used potentiometry, Read [66] conductivity, Izatt et al. [67] calorimetry and Rudolph [68] Raman spectroscopy techniques. At 373 K and above, the pKa,m values reported by Rudolph [68], Read [66] and Izatt et al. [67] are consistent with our results to within the combined experimental uncertainties. The results are also consistent with the potentiometric measurements from Mesmer and Baes [47] up to 450 K; but above this temperature the temperature dependence differs. The residuals in the fit, or differences between the experimental data and the fit under the same conditions (pKexp−pKfit), are shown in Fig. 4.b and reach up to ~ 0.3 pK units at 573 K. The cause of the discrepancy with the other studies observed at high temperature is not clear; Read [66] postulated the presence of polyphosphate species in Mesmer and Baes’ experimental solutions. The data from Mesmer and Baes were included in our fit (Eq. 26) which is able to represent their data up to 450 K to within their reported experimental uncertainties.
The fitted value given by Eq. 26 at 298 K and 0.1 MPa (pKa,m = 2.143 ± 0.016) is consistent with the Nuclear Energy Agency’s (NEA) recommended value for the first ionization constant of phosphoric acid in light water (pKa,m = 2.140 ± 0.030) [79, 80] and the more recent critical evaluation by Rard and Wolery [81]. These recommended values compare well with Read’s result [pKa,m(298.15, 0.1 MPa) = 2.15 ± 0.01], which was carefully estimated from his 20 MPa measurements [pKa,m(298.15, 20 MPa) = 2.09 ± 0.01] by considering the change in the partial molar volume of reaction (ΔrV°) for Eq. 11. Our result at 298 K and 20 MPa (pKa,m = 2.07 ± 0.02) is consistent with Read to within the combined experimental uncertainties and the pressure effect is similar to that for the ionization constant of light water at 298.15 K [pKw,m(0.1 MPa)−pKw,m(20 MPa) = 0.074] [39].
4.2.2 Acetic Acid pK a,m in H2O
The ionization constant of acetic acid has also been well-studied in H2O [18, 19, 48, 57, 58, 69,70,71,72,73,74,75,76,77,78, 82]. Briefly, Noyes et al. [69], MacInnes and Shedlovsky [70], La Mer et al. [58, 82], Ellis [71], Fisher and Barnes [72], Zimmerman and Wood [57], and Erickson et al. [18, 19] used conductivity, Harned and Ehlers [73], Schwarzenbach et al. [74], Glasoe and Long [75], Gary et al. [76], Mesmer et al. [48], Sue et al. [77] used potentiometry, and Oscarson et al. [78] used calorimetry techniques. Between 373 and 450 K, our results agree with the literature to within the combined experimental uncertainties. Above 450 K larger discrepancies appear, consistent with the difficulties with making hydrothermal measurements using different techniques. Except for Mesmer et al. [48] these remain to within ± 0.1 pK units, as shown by the residual plot in Fig. 5b. Similar to what was observed for H3PO4, the acetic acid potentiometric pK data from Mesmer et al. start to deviate from the other results around 450 K, showing up to 0.3 pK units difference at 573 K. It might be that there is a systematic error in the Oak Ridge National Laboratory (ORNL) results that becomes gradually more significant above 450 K due to assumptions in their data treatment. This matter was not addressed in their study and deserves closer attention. The data from Mesmer et al. were included in the fit (Eq. 26) weighted by the inverse square of their standard uncertainties.
The fitted value given by Eq. 26 at 298 K and 0.1 MPa (pKa,m = 4.764 ± 0.014) is consistent with MacInness and Shedlovsky’s value for the first ionization constant of acetic acid in light water (pKa,m = 4.756 ± 0.010) [70] and the other low temperature literature studies (e.g., [57, 73]). The pressure effect on our conductivity result [pKa,m(298.15 K,11.5 MPa) = 4.701 ± 0.014] is similar to that observed for the ionization constant of phosphoric acid.
4.3 D2O Isotope Effect ΔpK on the First Ionization Constant of Phosphoric Acid and the Ionization Constant of Acetic Acid
Experimental values of ΔpKa,m = [pKa,m(D2O)—pKa,m(H2O)] for phosphoric and acetic acid are listed in Tables 9 and 10 and plotted in Fig. 6 alongside literature results for phosphoric acid and acetic acid. To maintain consistency with our experimental approach to yield the most accurate difference in the ionization constant between light and heavy water, we chose to model the experimental values for ΔpKa,m directly [20, 21, 29] according to the expression:
where Δp1, Δp2, and Δp3 are adjustable fit parameters, T is the temperature in Kelvin, and \({V}_{{\mathrm{D}}_{2}\mathrm{O}}^{*}= {\mathrm{M}}_{{\mathrm{D}}_{2}\mathrm{O}}/{\rho }_{{\mathrm{D}}_{2}\mathrm{O}}\) is D2O molar volume in cm3·mol−1. The fitted parameters are given with their uncertainties in Tables 11 and 12. Combining these parameters with those for Eq. 26 from the light water data will yield the corresponding equation for heavy water, as given in Tables 11 and 12.

a Deuterium isotope effect on the ionization constant, ΔpKa,m, of phosphoric acid vs T. b Residuals: open star, [71]; open hexagon, [47]; open circle, [70]; open triangle, [60]; open diamond, [69]; filled circle, this work; solid line, Eq. 26 (p = 20 MPa); doted solid line, Eq. 26 (p = psat). Symbols in red were not included in the fit
No other groups have reported experimental data on ΔpK for any of these ionization reactions above 323 K. A few studies on the deuterium isotope effect on the first ionization constant of phosphoric acid have been reported [63,64,65, 68, 74], and except for Ref. [65], which reported \(\text{p}{K}_{\mathrm{a1},\mathrm{m}}{\text{D}_{3}\text{PO}_{4}}\) from T = 278 K to T = 323 K, all the studies focused on ambient conditions. Similar for observations can be made about the acetic acid system [18, 19, 58, 64, 75, 76], with the measurements reported from other groups also stopping at 323 K [76].
4.3.1 Selected Values for D3PO4and CH3COOD ΔpK a at 298.15 K
A few studies have reported ΔpKa values for D3PO4 and CH3COOD at 298 K. For D3PO4 Glasoe and Long reported ΔpKa,c = 0.20 (ΔpKa,m = 0.24) [75], McDougall and Long ΔpKa,c = 0.234 (ΔpKa,m = 0.278) [63], Paabo and Bates ΔpKa,m = 0.272 [65], Salomaa et al. ΔpKa,c = 0.205 (ΔpKa,m = 0.249) [64] and Rudolph ΔpKa,m = 0.280 [68]; for CH3COOD La Mer et al. reported ΔpKa,c = 0.514, (ΔpKa,m = 0.559) [58, 82], Glasoe and Long ΔpKa,m = 0.56 [75] and Gary et al. ΔpKa,m = 0.557 [76]. All these data were measured at 0.1 MPa. While the acetic acid ΔpKa,m data are in fair agreement, the phosphoric acid literature show some discrepancies. At 293 K the results from Schwarzenbach et al. [ΔpKa,m(D3PO4) = 0.249, ΔpKa,m(CH3COOD) = 0.499] [74] compare less well with Paabo and Bates [ΔpKa,m(D3PO4) = 0.181] [65] and Gary et al. [ΔpKa,m(CH3COOD) = 0.561] [76].
Our 298 K result at 20 MPa for D3PO4 (ΔpKa,c = 0.351, ΔpKa,m = 0.396) and CH3COOD (ΔpKa,c = 0.592, ΔpKa,m = 0.637) does not compare well with the other literature data at 0.1 MPa, with differences of about ~ 0.12 and ~ 0.08 pK units, respectively. The cause of the discrepancies between our conductivity results and the other literature data at 298 K is not clear and the effect seems too large to attribute to a pressure effect in going from 0.1 to 20 MPa. The change in the partial molar volume of reaction (ΔrV°) for the ionization of D3PO4 and CH3COOD were not investigated as part of this work and further studies are underway in our laboratory to quantify these effects. Paabo and Bates [65] did not study the deuterium isotope effect on ΔpKa,m(D3PO4) directly and instead made use of data from Bates [83] measured 19 years previously. They noted that some of the assumptions in their pKa calculations could have introduced “major uncertainty”. Salomaa et al. [64] also observed discrepancies up to 0.07 pK units between the results from their potentiometric and conductivity techniques.
ΔpKa,m experiments are difficult and because our flow conductivity instrument is designed for high temperature measurements it was decided not to anchor the fit discussed below to the 298 K data reported in this study. For phosphoric acid the 298 K value from Paabo and Bates (ΔpKa,m = 0.272) [65] was included, whereas for acetic acid all the literature sources at low temperature were included in the fit.
4.3.2 Equations of State for the First Ionization Constant of Phosphoric Acid and the Ionization Constant of Acetic Acid in D2O
The temperature and pressure dependence of the ionization of phosphoric and acetic acids in heavy water are given by:
where p1, p2, and p3 were calculated by adding the adjustable fit parameters for Eqs. 26 and 27. Parameters are tabulated in Tables 11 and 12 and the difference in ionization are shown in Figs. 7 and 8.

a Deuterium isotope effect on the ionization constant, ΔpKa,m, of acetic acid vs T. b Residuals: open star, [71]; open diamond, [35, 36]; addition sign, [26]; multiplication sign , [70]; open circle, [25]; open triangle, [21, 22]; filled circle, This Work; solid line, Eq. 26 (p = 20 MPa); doted solid line, Eq. 26 (p = psat). Symbols in red were not included in the fit

Acid ionization constants for a phosphoric and b acetic acid in H2O and D2O, pK vs T, from this work and literature. a open square, [50]; filled diamond, [56]; open hexagon, [47]; open circle, [70]; open triangle, [60]; open inverted triangle, [67]; filled square, [33]; open diamond, [69]; open star, [71] (not included in the fit); filled circle, This Work; solid line, Eq. 26 (p = 20 MPa); –-, Eq. 26 (p = psat); b open triangle, [21, 22]; open square, [52]; open hexagon, [81]; open diamond, [35, 36]; open inverted triangle, [57] recalculated by [81]; addition sign , [43]; filled inverted triangle, [20]; filled triangle, [24]; multiplication sign , [29]; open star, [71]; filled diamond, [26]; open circle, [25]; filled square, [73]; filled hexagon, [59]; filled circle, This Work; solid line, Eq. 26 (p = 20 MPa); doted solid line, Eq. 26 (p = psat)
5 Conclusions
This work reports accurate ΔpKa,m measurements of the first ionization of phosphoric acid between 373 and 573 K. These are the first experimental D3PO4 ionization constants reported above 323 K. The results are consistent with other literature studies to within the combined experimental uncertainties and confirm that the supercritical flow AC conductivity cell is a powerful tool to measure ionization constants and the D2O isotope effect under hydrothermal conditions. Observed differences up to 0.3 pK units in pKa,m for both phosphoric and acetic acid in H2O from Mesmer et al. [47, 48] suggest that there might be a systematic error in the ORNL treatment resulting in an increasing systematic error above 450 K. Discrepancies with other studies observed at 298 K were not resolved as part of this work and will be addressed in a future publication. The ΔpKa,m results reported here complete a series of experimental projects on acid ionization started in our research group [16,17,18,19,20,21,22] and together the results of these studies provide the basis for developing a predictive model for transition metal hydrolysis and oxide solubility in D2O under CANDU-PHW primary coolant conditions, from H2O-based measurements. We are exploring the use of DFT methods [7] to seek a better understanding of the differences in ionic hydration between light and heavy water systems, and these results together with the new-built high temperature D2O database will be used to derive algorithms for computer codes used by the nuclear industry to calculate and monitor chemical speciation and pH in heavy water coolant and moderator conditions such as the EPRI code MULTEQ [84].
References
Cook, W.G., Lister, D.H.: Chemistry in CANDU Process Systems (Chapter 15). In: Wm. J. Garland. (ed.) The Essential CANDU: A textbook on the CANDU Nuclear Power Plant Technology. UNENE, Hamilton (2014)
Palmer, D.A., Fernández-Prini, R., Harvey, A.H.: Aqueous Systems at Elevated Temperatures and Pressures—Physical Chemistry in Water Steam and Hydrothermal Solutions. Elsevier Ltd., Amsterdam (2004)
Valyashko, V.: Hydrothermal Properties of Materials: Experimental Data on Aqueous Phase Equilibria and Solution Properties at Eelevated Temperatures and Pressures. Wiley, Hoboken (2008)
Brown, P.L., Ekberg, C.: Hydrolysis of Metal Ions. Wiley, Hoboken (2016)
Arnett, E.M., McKelvey, D.R.: Solvent isotope effect on thermodynamics of non-reacting solutes. In: Cotezee, J.F., Ritchie, C.D. (eds.) Solute-Solvent Interactions. Marcel Dekker, NewYork (1969)
Bates R. G.: Solute-solvent interactions, Chapter 4. In: Coetzee, J. F., Ritchie, C. D., (eds.). Marcel Dekker, New York (1969)
Conrad, J. K.: Deuterium Isotope Effects on Acid Ionization and Metal Oxide Hydrolysis Under Hydrothermal Conditions. Ph.D. Thesis, University of Guelph, Guelph, (2020)
Hassanali, A., Gibertia, F., Cuny, J., Kühnec, T.D., Parrinello, M.: Proton transfer through the water gossamer. Proc. Nat. Acad. Sci. U S A 110, 13723–13728 (2013)
Marx, D.: Proton transfer 200 years after von Grotthuss: insights from Ab initio simulations. Chem. Phys. Chem. 7, 1848–1870 (2006)
Mora-Diez, N., Egorova, Y., Plommer, H., Tremaine, P.R.: Theoretical study of deuterium isotope effects on acid-base equilibria under ambient and hydrothermal conditions. RSC Adv. 2, 9097–9109 (2015)
Conrad, J.K., Tremaine, P.R.: A Study of the deuterium isotope effect on zinc(II) hydrolysis and solubility under hydrothermal conditions using density functional theory. Chem. Eng. Sci. 254, 117596–117602 (2022)
Laughton, P.M., Robertson, R.E.: Solvent isotope effects for equilibria and reactions. In: Coetzee, J.F., Ritchie, C.D. (eds.) Solvent Interactions in Solute-Solvent Interactions. Marcel Dekker, New York (1969)
Lietzke, M.H., Stoughton, R.W.: The second dissociation constant of deuterosulfuric acid from 25 to 225°C. J. Phys. Chem. 67, 652–654 (1963)
Shoesmith, D.W., Lee, W.: The ionization of heavy water (D2O) in the temperature range 298 to 523 K. Can. J. Chem. 54, 3553–3558 (1976)
Mesmer, R.E., Herting, D.L.: Thermodynamics of ionization of D2O and D2PO4-. J. Solution Chem. 7, 901–913 (1978)
Trevani, L.N., Balodis, E., Tremaine, P.R.: Apparent and standard partial molar volumes of NaCl, NaOH, and HCl in water and heavy water at 523 K and 573 K at p = 14 MPa. J. Phys. Chem. B 111, 2015–2024 (2007)
Bulemela, E., Tremaine, P.R.: D2O isotope effects on the ionization of β-naphthol and boric acid at temperatures from 225 to 300 °C using UV-Visible Spectroscopy. J. Solution Chem. 38, 805–826 (2009)
Erickson, K.M., Arcis, H., Raffa, D., Zimmerman, G.H., Tremaine, P.R.: Deuterium isotope effects on the ionization constant of acetic acid in H2O and D2O by AC conductance from 368 to 548 K at 20 MPa. J. Phys. Chem. B 115, 3038–3151 (2011)
Erickson, K.M., Arcis, H., Raffa, D., Zimmerman, G.H., Tremaine, P.R.: Correction to “deuterium isotope effects on the ionization constant of acetic acid in H2O and D2O by AC Conductance from 368 to 548 K at 20 MPa.” J. Phys. Chem. B 123, 9503–9506 (2019)
Conrad, J.K., Sasidharanpillai, S., Tremaine, P.R.: Second dissociation constant of carbonic acid in H2O and D2O from 150 to 325°C at p = 21 MPa using Raman Spectroscopy and a sapphire-windowed flow cell. J. Phys. Chem. B 124, 2600–2617 (2020)
Conrad, J.K., Tremaine, P.R.: Third dissociation constant of phosphoric acid in H2O and D2O from 75 to 300°C at p = 20.4 MPa using Raman spectroscopy and a Titanium sapphire flow cell. Phys. Chem. Chem. Phys. 23, 10670–10685 (2021)
Yacyshyn, M.B., Applegarth, L.M.S.G.A., Cox, J.S., Tremaine, P.R.: Deuterium isotope effects on the second ionization constant of aqueous sulfuric acid from 25°C to 200°C using Raman Spectroscopy. J. Solution Chem. 51, 479–498 (2022)
Zimmerman, G.H., Gruskiewicz, M.S., Wood, R.H.: New apparatus for conductance measurements at high temperatures: conductance of aqueous solutions of LiCl, NaCl, NaBr, and CsBr at 28 MPa and water densities from 700 to 260 kg m-3. J. Phys. Chem. 99, 11612–11625 (1995)
Zimmerman, G.H., Arcis, H., Tremaine, P.R.: Limiting conductivities and ion association constants of aqueous NaCl under hydrothermal conditions: experimental data and correlations. J. Chem. Eng. Data 57, 2415–2429 (2012)
Fernández-Prini, R.: Conductance of electrolyte solutions. A modified expression for its concentration dependence. Trans. Faraday Soc. 65, 3311–3313 (1969)
Quint, J., Viallard, A.: Relaxation field for the general case of electrolyte mixtures. J. Solution Chem. 7, 137–153 (1978)
Quint, J., Viallard, A.: The Electrophoretic effect for the case of electrolyte mixtures. J. Solution Chem. 7, 525–531 (1978)
Quint, J., Viallard, A.: Electrical conductance of electrolyte mixtures of any type. J. Solution Chem. 7, 533–548 (1978)
Conrad, J.K., Arcis, H., Ferguson, J.P., Tremaine, P.R.: Second ionization constant of sulfuric acid in H2O and D2O from 150 to 300°C at p = 11.5 MPa using flow AC conductivity. Phys. Chem. Chem. Phys. 25, 1659–1676 (2023)
Arcis, H., Zimmerman, G.H., Tremaine, P.R.: Ion-pair formation in aqueous strontium chloride and strontium hydroxide solutions under hydrothermal conditions by AC conductivity measurements. Phys. Chem. Chem. Phys. 16, 17688–17704 (2014)
Arcis, H., Ferguson, J.P., Zimmerman, G.H., Tremaine, P.R.: The limiting conductivity of the borate ion and its ion-pair formation constants with sodium and potassium under hydrothermal conditions. Phys. Chem. Chem. Phys. 18, 24081–24094 (2016)
Arcis, H., Ferguson, J.P., Applegarth, L.M.S.G.A., Zimmerman, G.H., Tremaine, P.R.: Ionization of boric acid in water from 298 K to 623 K by AC conductivity and Raman Spectroscopy. J. Chem. Thermodyn. 106, 187–198 (2017)
Arcis, H., Ferguson, J.P., Cox, J.S., Tremaine, P.R.: The ionization constant of water at elevated temperatures and pressures new data from direct conductivity measurements and revised direct conductivity measurements and revised formulations from T = 273 K to 674 K and p = 0.1 MPa to 31 MPa. J. Phys. Chem. Ref. Data (2020). https://doi.org/10.1063/1.5127662
Plumridge, J., Arcis, H., Tremaine, P.R.: Limiting conductivities of univalent cations and the chloride ion in H2O and D2O under hydrothermal conditions. J. Solution Chem. 44, 1062–1089 (2015)
Zimmerman, G.H., Arcis, H.: Extrapolation methods for AC impedance measurements made with a concentric cylinder cell on solutions of high ionic strength. J. Solution Chem. 44, 912–933 (2015)
Zimmerman, G.H., Arcis, H., Tremaine, P.R.: Limiting conductivities and ion association in aqueous NaCF3SO3 and Sr(CF3SO3)2 from 298 to 623 K at 20 MPa. Is triflate a non-complexing anion in high temperature water? J. Chem. Eng. Data 57, 3180–3197 (2012)
Marshall, W.L.: Reduced state relationship for limiting electrical conductances of aqueous ions over wide ranges of temperature and pressure. J. Chem. Phys. 87, 3639–3643 (1987)
Marshall, W.L., Franck, E.U.: Ion product of water substance, 0–1000 °C, 1–10,000 bars New International Formulation and its background. J. Phys. Chem. Ref. Data 10, 295–304 (1981)
Bandura, A.V., Lvov, S.N.: The ionization constant of water over wide ranges of temperature and density. J. Phys. Chem. Ref. Data 35, 15–30 (2006)
E. W. Lemmon, I. H. Bell, M. L. Huber and M. O. McLinden, NIST standard reference database 23: reference fluid thermodynamic and transport properties-REFPROP, Version 10.0, Gaithersburg: National Institute of Standards and Technology (Standard Reference Data Program), 2018.
Bianchi, H.L., Dujovne, I., Fernández-Prini, R.: Comparison of electrolytic conductivity theories: performance of classical and new theories. J. Solution Chem. 29, 237–253 (2000)
Turq, P., Blum, L., Bernard, O., Kunz, W.: Conductance in associated electrolytes using the mean spherical approximation. J. Phys. Chem. 99, 822–827 (1995)
Lee, W.H., Wheaton, R.J.: Conductance of symmetrical, unsymmetrical and mixed electrolytes. Part 1.–Relaxation terms. J. Chem. Soc. Faraday Trans. 2 74, 743–766 (1978)
Lee, W.H., Wheaton, R.J.: “Conductance of symmetrical, unsymmetrical and mixed electrolytes. Part 2.–Hydrodynamic terms and complete conductance equation. J. Chem. Soc. Faraday Trans. 2 74, 1456–1482 (1978)
Lee, W.H., Wheaton, R.J.: “Conductance of symmetrical, unsymmetrical and mixed electrolytes. Part 3.–Examination of new model and analysis of data for symmetrical electrolytes. J. Chem. Soc. Faraday Trans. 2 75, 1128–1145 (1979)
Johnson, J.W., Oelkers, E.H., Helgeson, H.C.: SUPCRT92: a software package for calculating the standard Molal thermodynamic properties of minerals, gases, aqueous species, and reactions from 1 to 5000 bar and 0 to 1000°C. Comput. Geosci. 18, 899–947 (1992)
Mesmer, R.E., Baes, C.F., Jr.: Phosphoric acid dissociation equilibria to 300°C. J. Solution Chem. 3, 307–322 (1974)
Mesmer, R.E., Patterson, C.S., Busey, R.H., Holmes, H.F.: Ionization of acetic acid in aq. sodium chloride media: a potentiometric study to 573K and 130 bar. J. Phys. Chem. 93, 7483–7490 (1989)
Okada, K., Yao, M., Hiejima, Y., Kohno, H., Kajihara, Y.: Dielectric relaxation of water and heavy water in the whole fluid phase. J. Chem. Phys. 110, 3026–3036 (1999)
Uematsu, M., Frank, E.U.: Static dielectric constant of water and steam. J. Phys. Chem. Ref. Data 9, 1291–1306 (1980)
Tsurko, E.N., Neueder, R., Barthel, J., Apelblat, A.: Conductivity of phosphoric acid, sodium, potassium, and ammonium phosphates in dilute aqueous solutions from 278.15 K to 308.15 K. J. Solution Chem. 28, 973–999 (1999)
Robinson, R.A., Stokes, R.H.: Electrolyte Solutions (Revised 2nd Edition), Mineola. Dover Publications Inc, New York (2002)
Muccitelli, J.A., DiAngelo, N.A.: Electrolytic conductivity of aqueous solutions of potassium and sodium phosphates to 325°C. J. Chem. Eng. Data 39, 131–133 (1994)
Arcis, H., Plumridge, J., Tremaine, P.R.: Limiting conductivities of strong acids and bases in D2O and H2O: deuterium isotope effects on proton hopping over a wide temperature range. J. Phys. Chem. B 43, 8791–8803 (2022)
Ho, P.C., Palmer, D.A., Wood, R.H.: Conductivity measurements of dilute aqueous LiOH, NaOH, and KOH solutions to high temperatures and pressures using a flow-through cell. J. Phys. Chem. B 104, 12084–12089 (2000)
Ho, P.C., Palmer, D.A., Gruszkiewicz, M.S.: Conductivity measurements of dilute aqueous HCl solutions to high temperatures and pressures using a flow-through cell. J. Phys. Chem. 105, 1260–1266 (2001)
Zimmerman, G.H., Wood, R.H.: Conductance of dilute sodium acetate solutions to 469 K and of acetic acid and sodium acetate/acetic acid mixtures to 548 K and 20 MPa. J. Solution Chem. 31, 995–1017 (2002)
La Mer, V.K., Chittum, J.P.: The conductance of salts (potassium acetate) and the dissociation constant of acetic acid in deuterium oxide. J. Am. Chem. Soc. 58(9), 1642–1644 (1936)
Brescia, F., LaMer, V.K., Nachod, F.C.: The temperature dependence of the dissociation constant of deuteroacetic acid. J. Am. Chem. Soc. 62(3), 614–617 (1940)
Méndez de Leo, L., Wood, R.H.: Conductance study of association in aqueous CaCl2, Ca(CH3COO)2, and Ca(CH3COO)2·nCH3COOH from 348 to 523 K at 10 MPa. J. Phys. Chem. B 109(29), 14243–14250 (2005)
Mesmer, R.E., Marshall, W.L., Palmer, D.A., Simonson, J.M., Holmes, H.F.: Thermodynamics of aqueous association and ionization reactions at high temperatures and pressures. J. Solution Chem. 17, 699–718 (1988)
Nims, L.F.: The first dissociation constant of phosphoric acid from 0 to 50°C. J. Am. Chem. Soc. 56(5), 1110–1112 (1934)
McDougall, A.O., Long, F.A.: Relative hydrogen bonding of deuterium. II. acid ionization constants in H2O and D2O. J. Phys. Chem. 66, 429–433 (1962)
Salomaa, P., Schaleger, L.L., Long, F.A.: Solvent deuterium isotope effects on acid-base equilibria. J. Am. Chem. Soc. 86(1), 1–7 (1964)
Paabo, M., Bates, R.G.: Deuterium isotope effects and the dissociation of deuteriophosphoric acid from 5 to 50°C. J. Phys. Chem. 74, 706–710 (1970)
Read, A.J.: The first ionization constant from 25 to 200°c and 2000 bar for orthophosphoric acid. J. Solution Chem 17, 213–224 (1988)
Izatt, R.M., Gillespie, S.E., Oscarson, J.L., Wang, P., Renuncio, J.A.R., Pando, C.: The Effect of temperature and pressure on the protonation of o-phosphate ions at 348.15 and 398.15 K, and at 1.52 and 12.50 MPa. J. Solution Chem. 23, 449–468 (1994)
Rudolph, W.W.: Raman- and infrared-spectroscopic investigations of dilute aqueous phosphoric acid solutions. Dalton Trans. 39, 9642–9653 (2010)
Noyes, A.A., Melcher, A.C., Cooper, H.C., Eastman, G.W., Kato, Y.: The conductivity and ionization of salts, acids, and bases in aqueous solutions at high temperatures. J. Am. Chem. Soc. 30(3), 335–353 (1908)
MacInnes, D.A., Shedlovsky, T.: The determination of the ionization constant of acetic acid at 25°C, from conductance measurments. J. Am. Chem. Soc. 54(4), 1429–1438 (1932)
Ellis, A.J.: The Ionization of acetic, propionic, n-butyric, and benzoic acid in water, from conductance measurements up to 225°C. J. Chem. Soc. 2299–2310 (1963)
Fisher, J.R., Barnes, H.L.: The Ion-product constant of water to 350°C. J. Phys. Chem. 76(1), 90–99 (1972)
Harned, H.S., Ehlers, R.W.: The dissociation constant of acetic acid from 0 to 60°C. J. Am. Chem. Soc. 55(2), 652–656 (1933)
Schwarzenbach, G., Epprecht, A., Erlenmeyer, H.: Uber Dissoziationskonstanten in Wasser und Deuteriumoxyd. Messungen mit der Deuteriumelektrode. Helvetica 19(1), 1292–1304 (1936)
Glasoe, P.K., Long, F.A.: Oxide, use of glass electrodes to measure acidities in deuterium. J. Phys. Chem. 64(1), 188–190 (1960)
Gary, R., Bates, R.G., Robinson, R.A.: Dissociation Constant of acetic acid in deuterium oxide from 5 to 50°C. Reference points for a pD scale. J. Phys. Chem. 69(8), 2750–2753 (1965)
Sue, K., Usami, T., Arai, K.: Determination of acetic acid dissociation constants to 400 °C and 32 MPa by potentiometric pH measurements. J. Chem. Eng. Data 48, 1081–1084 (2003)
Oscarson, J.L., Gillespie, S.E., Christensen, J.J., Izatt, R.M., Brown, P.R.: Thermodynamic quantities for the interaction of H+ and Na+ with C2H3O −2 and Cl− in aqueous solution from 275 to 320°C. J. Solution Chem. 17(9), 865–885 (1988)
Grenthe, I., Fuger, J., Konings, R.J., Lemire, R.J., Muller, A.B., Nguyen-Trung, C., Wanner, H.: Chemical Thermodynamics of Uranium, vol. 1. Elsevier, Amsterdam (1992)
Guillaumont, R., Mompean, F.J.: Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium, vol. 5. Elsevier, Amsterdam (2003)
Rard, J.A., Wolery, T.J.: The standard chemical-thermodynamic properties of phosphorus and some of its key compounds and aqueous species: an evaluation of differences between the previous recommendations of NBS/NIST and CODATA. J. Solution Chem. 36, 1585–1599 (2007)
Korman, S., La Mer, V.K.: Deuterium exchange equilibria in solution and the quinhydrone electrode. J. Am. Chem. Soc. 58(8), 1396–1403 (1936)
Bates, R.G.: First dissociation constant of phosphoric acid from 0 to 60C; limitations of the electromotive force method for moderately strong acids. J. Res. Nat. Bur. Stand. 47(3), 127–134 (1951)
“MULTEQ Version 9.0 (2018): Equilibrium of an electrolytic solution with vapor–liquid partitioning and precipitation—The database,” EPRI, Palo Alto, CA: 3002013656
Acknowledgements
The authors are grateful to Prof. R.H. Wood (University of Delaware), Prof. G.H. Zimmerman (Bloomsburg University) for many suggestions and technical discussions related to the experimental work. Technical advice and encouragement were provided by Dr. Jenny Cox. Mr. Ian Renaud and Mr. Case Gielen provided capable electronic and machine-shop support, particularly in reconstructing the conductivity flow cell. The D2O purity analysis was completed using the NMR Center at the University of Guelph. Scientific advice and encouragement were also provided by Dr. Dave Guzonas, Atomic Energy of Canada Ltd.; Dr. Dave Evans, Ontario Power Generation Ltd. (OPG); and Dr. Mike Upton, Bruce Power Ltd. Ontario Power Generation Inc. supplied the heavy water used in this work. This work was originally funded by the Natural Sciences and Engineering Council of Canada (NSERC) and the University Network of Excellence in Nuclear Engineering (UNENE) as NSERC Collaborative Research Grants CRDPJ 381418 – 09 and CRDPJ 447964 – 13. This research was carried out under the NSERC/UNENE Senior Industrial Research Chair Program at the University of Guelph, which is supported by the Natural Science and Engineering Research Council of Canada (NSERC Grant IRCPJ 499567-15), and the University Network of Excellence in Nuclear Engineering (UNENE), the CANDU Owners Group (COG), the Nuclear Waste Management Organization (NWMO), and the Electric Power Research Institute (EPRI).
Author information
Authors and Affiliations
Contributions
The 20 MPa phosphoric and acetic acid studies were carried out as part of the graduate program of KME (Ph.D. Thesis, University of Guelph, 2013). HA carried out a significant reanalysis of the data reported in the PhD thesis. JPF contributed to the development of the methodology used in more recent measurements at 11.5 MPa, carried out the experiments, and contributed to the data analysis. JKC assisted with the 11.5 MPa data collection, performed most of the data analysis with the FHFP and QV conductivity models, density model fits, and preparation of Figures and Tables. The work carried out under the direction of PRT and HA. The manuscript was written by HA and PRT with contributions from JPF and JKC.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Arcis, H., Conrad, J.K., Ferguson, J.P. et al. First Ionization Constant of Phosphoric Acid and of Acetic Acid in H2O and D2O from T = 373 K to 573 K at p = 11.5 and 20 MPa by AC Conductivity Methods. J Solution Chem 53, 91–125 (2024). https://doi.org/10.1007/s10953-023-01281-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-023-01281-4