
Abstract
O-Phospho-l-serine is one of the naturally occurring phosphorylated amino acids, having important pharmacological activity and bioactivity. The protonation constants of O-phospho-l-serine were determined by means of potentiometric titrations at 25 °C and ionic strength of 0.5 mol·L−1 (NaCl). The heat effects of the protonation reaction of the O-phospho-l-serine were measured by direct calorimetry. NMR spectroscopy has demonstrated that the first protonation site occurs at the nitrogen atom in the amino group, followed by one of the oxygen atoms in the phosphono group, and finally the carboxyl oxygen atom. This trend is in good agreement with the enthalpy of protonation and quantum chemical calculations. These data will help to predict the speciation of O-phospho-l-serine in physiological systems.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
One of the most important processes of protein modification and signal transmission in cells is phosphorylation. It is estimated that about 30% of proteins encoded by the human genome contain covalently bound phosphate [1, 2]. Reversible protein phosphorylation, principally on serine, threonine and tyrosine residues, is one of the most important and best-studied post-translational modifications. Phosphoserine is a component of many proteins as the result of post-translational modifications. The phosphorylation of the alcohol functional group in serine to produce phosphoserine is catalyzed by various types of kinases. Through the use of technologies that utilize an expanded genetic code, phosphoserine can also be incorporated into proteins during translation [3]. The structural characterization of phosphoserine is a subject of considerable interest due to its importance in physiological systems.
Phosphoserine has three potential coordination sites (carboxyl, amine and phosphate group) to coordinate in various fashions under different reaction conditions [4,5,6]. Determination of the mode of coordination between ligand and metal ions occurring in an organism is a first step in explaining the function of phosphoserine in bioinorganic processes. Complex formation of phosphoserine with metal ions depends on the pH of the reaction medium, i.e. the degree of protonation. Protonation reactions are very important for biologically active molecules because of their ability to switch the molecule from proton-acceptor to proton-donor with only small changes in the pH environment. Protonation of phosphoserine may play a crucial role in enzymatic chemistry. Knowledge of the thermodynamics of the protonation of phosphoserine possibly offers insight into understanding how phosphorproteins can be regulated in physiological systems.
To study the mechanisms of biochemical reactions, it is necessary to know as accurately as possible the speciation. In order to have a complete picture of the speciation of phosphoserine, investigations of its protonation reactions have been performed by a few authors [7,8,9,10,11,12]. However, thermodynamic parameters other than stability constants, e.g., enthalpy of complex formation, are rarely available. As a result, it is difficult to predict the chemical behavior in solution. Our group is especially interested in thermodynamic studies because of their importance in providing fundamental information on the nature (e.g., ionic bonding versus covalence bonding, outer sphere versus inner sphere), energetics (e.g., Gibbs energy, enthalpy, entropy and heat capacity), structures and stabilities of complexes, and this paper presents a thermodynamic study on the protonation reaction of O-phospho-l-serine.
2 Experimental
2.1 Chemicals
All chemicals were reagent grade or higher. Milli-Q water was used for all analytical work described in this paper. O-Phospho-l-serine was purchased from Sigma–Aldrich. The purity was assumed to be 100% and checked by alkalimetric titration. NaCl (≥ 98%, Aladdin) was chosen as the background electrolyte. A stock solution of O-phospho-l-serine (H3L) was prepared by dissolving the desired amount of solid in a solution containing 0.5 mol·L−1 NaCl. The Milli-Q water was boiled in quartz glassware to remove dissolved CO2 to prepare the NaOH solution. Working solutions of NaOH and HCl containing NaCl were standardized by titration with potassium hydrogen phthalate (primary standard, Alfa Aesar) and Trizma base (crystalline, Sigma), respectively. The carbonate content of the NaOH solution was confirmed to be < 1% using a Gran’s plot [13, 14]. The ionic strength of all working solution was maintained at 0.5 mol·L−1 (NaCl). Deuterium oxide (99.8% D, J&K) was used to prepare the NMR sample solution.
2.2 Potentiometry
The instrument setup involved a potentiometric titrator (888 Titrando, Metrohm) equipped with a combination pH electrode (Orion™ 8102BN ROSS™). A circulating water-jacketed titration cell (100 mL) was used to maintain a constant temperature at 25.0 ± 0.1 °C. Solutions for experiments were protected by argon throughout the titration to avoid the contamination by carbon dioxide. Titrations of HCl with NaOH solution to obtain the electrode parameters, which allowed the calculation of hydrogen ion concentrations from the electrode potential in the subsequent titration conducted at the same temperature and ionic strength (Gran’s method) [13, 14]. To minimize systematic errors and to check the repeatability of the measurements, multiple titrations were conducted with solutions containing different concentration of the ligand (C L as total ligand concentration) and acidity (C H for total hydrogen ion). Usually, about 50 points were collected for each titration. The protonation constants (log10 β) were resolved by the nonlinear regression program (Hyperquad 2013) [15]. The 95% probability value is given in the program output.
2.3 Microcalorimetry
Microcalorimetric titrations at 25.0 ± 0.1 °C were performed on an isothermal titration microcalorimeter (ITC, TAM-III, TA Instruments) equipped with a nanocalorimeter and an automated titration syringe (Hamilton, 250 μL) to obtain the reaction enthalpy values for the protonation of O-phospho-l-serine in 0.5 mol·L−1 NaCl as described previously [16, 17]. Both chemical and electrical calibrations were performed to validate the performance of the instrument. The cell contained 750 µL O-phospho-l-serine (~ 5 µmol·L−1) stirred by a gold propeller maintained at 80 rpm. The syringe contained 250 μL NaOH (~ 50 mmol·L−1) titrant, which was delivered in 5 μL aliquots with a delay of 15 min between injections. The system was allowed to equilibrate before starting the titration experiments until a very stable baseline signal was obtained. Multiple titrations using different ligand concentrations were performed to reduce the uncertainty of the results. The dilution heat (Q dil) was then measured by titrating 750 µL of 0.5 mol·L−1 NaCl solution with the NaOH in the absence of O-phospho-l-serine. Therefore, the heat involved in deprotonation reaction of O-phospho-l-serine (Q corr) is: Q corr = Q meas − Q dil. The observed reaction heat is a function of a number of parameters, including the concentrations of reactants, the protonation constants (log10 β) and the enthalpy of protonation of the ligand (ΔH). Using the stoichiometric concentrations of the reactants and the protonation constants measured by potentiometry in this work, the enthalpies of protonation of O-phospho-l-serine at 25 °C were calculated from the ITC data by means of the computer program HypDeltaH [18].
2.4 NMR Spectroscopy
NMR experiments were performed on a 400 MHz Bruker Avance spectrometer. O-Phospho-l-serine was dissolved in D2O. NaOH in D2O was used to adjust the dominant speciation of the molecule in solution based on the protonation constants in aqueous solution. The sample was transferred to a standard NMR capillary and measured at room temperature. 1H and 13C NMR spectra were measured with DSS (sodium salt of 4,4-dimethyl-4-silapentane-1-sulfonic acid) as an internal standard. 31P NMR spectra were recorded with 85% H3PO4 as an external standard.
2.5 Quantum Chemical Calculations
The electron correlation effects were considered by employing density functional theory (DFT) methods at the ωB97X-D level of theory that included long-range corrections and empirical dispersion corrections [19,20,21]. Dunning’s AUG-cc-pVDZ basis set was used to describe the C, H, O, N and P atoms. All of species were optimized in aqueous medium while using the Truhlar and coworkers’ SMD solvation model [22, 23]. The default fine grid (75, 302), having 75 radial shells and 302 angular points per shell, was used to evaluate the numerical integration accuracy. The natural atomic charges and Wiberg bond indices (WBIs) were calculated on the basis of natural bond orbital (NBO) theory [24,25,26,27]. The WBIs is a measure of the bond order and, hence, of the bond strength between these two atoms [28]. All calculations were carried out using the Gaussian 09 program [29].
3 Results and Discussion
3.1 Protonation Constants of O-Phospho-l-serine
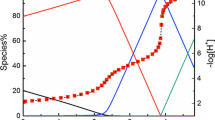
Representative potentiometric titrations of O-phospho-l-serine at 25 °C and the fitting curves are shown in Fig. 1. There are three successive protonation reactions in the pH range of this study. The fourth protonation reaction, on the second oxygen atom of the phosphonate group, occurs outside the pH range of the potentiometry, so it is not considered here. The speciation occurring during the course of the titration experiment is superimposed on these plots. The protonation constants (log10 β) of the molecule were calculated and are shown in Table 1. They are comparable to the constants reported in the literature. As shown in Table 1, all three protonation constants decrease with increasing temperature at ionic strength of 0.1 mol·L−1. This trend is in good agreement with the enthalpy of protonation directly determined by calorimetry. At 25 °C, from the results obtained, it seems that ionic strength is also an important factor in the apparent equilibrium constant.

Potentiometric titrations of the protonation of O-phospho-l-serine at 25 °C. I = 0.5 mol·L−1 NaCl. Initial solution: V 0 = 22.31 mL, \( {n_{{{\text{L}}^{3 - }}}} \) = 0.142 mmol, \( {n_{{{\text{H}}^+ }}} \) = 0.426 mmol; titrant: 48.35 mmol·L−1 NaOH (H3L denotes the neutral O-phospho-l-serine in solution)
3.2 NMR Spectroscopy
1H and 31P NMR spectra of four O-phospho-l-serine solutions (solutions A, B, C, and D) were collected to help identify the protonation sites (Figs. 2 and 3). Values of acidity (−log[H+ + D+], calculated by the Hyss2009 program based on stoichiometric concentrations of the reactants [31]) of the solutions are 11.495, 7.882, 3.710, and 1.411, for solutions A, B, C, and D, respectively. The dominant species of O-phospho-l-serine in these solutions were calculated to be: solution A: L3− (98.7%), solution B: HL2− (97.9%), solution C: H2L− (96.2%), and solution D: H3L (82.2%). Therefore, the variations in the NMR spectra from solution A to D can be discussed as reflecting the stepwise protonation reaction of O-phospho-l-serine.

1H NMR spectra of four O-phospho-l-serine solutions. The acidities (−log10[D+ + H+]) of the solutions are (A) 11.495, (B) 7.882, (C) 3.710, and (D) 1.411, respectively. The dominant species in these solutions were calculated to be: solution (A): L3− (98.7%), solution (B): HL2− (97.9%), solution (C): H2L− (96.2%), and solution (D): H3L (82.2%)

pH dependence of the 31P NMR phosphorus shift. The acidities (−log10[D+ + H+]) of the solutions are (A) 11.495, (B) 7.882, (C) 3.710, and (D) 1.411, respectively. The dominant species in these solutions were calculated to be: solution (A): L3− (98.7%), solution (B): HL2− (97.9%), solution (C): H2L− (96.2%), and solution (D): H3L (82.2%)
Before analyzing the 1H NMR spectra, the peaks in the spectra need to be assigned. The assignments of peaks in the 1H NMR spectra were accomplished with help from the 2D 13C–1H HSQC data. Usually, the largest chemical shift of 1H NMR spectra occurs closest to the site of protonation. As shown in Fig. 2, in going from solution A to B, the dominant species changes from L3− to HL2−, representing the first protonation step (H+ + L3− ⇋ HL2−). The largest chemical shift occurring on the hydrogen atoms b suggests that the first protonation occurs on the amine nitrogen atom.
31P NMR spectroscopy is a method well suited for the analysis of phosphorus-containing ligands because the 31P chemical shift is very sensitive to the phosphate’s state [32,33,34]. Prontonation of the phosphonate oxygen atom will alter electronegativity of the phosphorus atom and O–P–O bond angle [35, 36]. Then, a significant 31P chemical shift will be observed. As shown in Fig. 3, in going from solution B to C, the dominant species changes from HL2− to H2L−, representing the second protonation step (H+ + HL2− ⇋ H2L−). The largest 31P NMR chemical shift suggests the second protonation occurs on the phosphonate oxygen atom.
It is well known that H3L is in the zwitterionic form with one proton removed from the phosphate group and an extra one located on the amino nitrogen [37,38,39,40,41]. Although, the solid-state conformations may not exactly correspond to those in the aqueous environments of biology, molecular modeling indicates the preference of biologically active structures. By comparison of crystal structures as well as solution NMR studies, the successive protonation sites of the totally deprotonated molecule should be written as in Scheme 1. This trend is in agreement with the results from solid-state NMR spectroscopy and Raman spectroscopy in the literature on the subject [35, 42].

O-phospho-l-serine in its different ionized forms
3.3 Density Functional Theory (DFT) Calculations
In order to confirm that the first protonation reaction of fully deprotonated O-phospho-l-serine starts with amine nitrogen atom rather than with one of the oxygen atoms in the phosphono group, the structures of three possible sites (the phosphonate, the amine, and the carboxyl) in aqueous medium were calculated by the DFT methods. The three optimized structures are shown in Fig. 4. As shown in Fig. 4d, the order of the relative energies for the three situations is H–L(N) < H–L(OP) < H–L(OC) which indicates that H–L(N) is the most stable structure. In addition, as shown in Table 2, the magnitude of WBIs is also H–L(N) > H–L(OP) > H–L(OC), which corresponds to the results of energy calculations. These calculations show that the first protonation reaction occurs on the amine group.

The optimized structures of the stationary points for the various structures of the first protonation (HL2−) by DFT calculations: a H–L(amine), b H–L(phosphonate) and c H–L(carboxyl). The H–L bond lengths are shown in this figure. The relative energies for various HL2− are depicted in (d) which corresponds to the three optimized structures in (a), (b) and (c), respectively
3.4 Enthalpy of Protonation of O-Phospho-l-serine
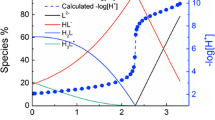
Isothermal titration calorimetry (ITC) is a technique to measure the stoichiometry and thermodynamics from binding experiments. Data of the calorimetric titrations for the protonation of O-phospho-l-serine are shown in Fig. 5. The enthalpies of protonation of O-phospho-l-serine at 25 °C are also presented in Table 1. The cumulative enthalpies of protonation measured by microcalorimetry at 25 °C are − 38.7 ± 0.4 kJ·mol−1 (for H+ + L3− ⇋ HL2−), − 38.8 ± 0.5 kJ·mol−1 (for 2H+ + L2− ⇋ H2L−), and − 40.0 ± 2.0 kJ·mol−1 (for 3H+ + L− ⇋ H3L), indicating that the cumulative three protonation steps are exothermic.

Calorimetric titrations of protonation of O-phospho-l-serine at 25 °C, I = 0.5 mol·L−1 NaCl: a thermogram (solid line, using bottom x-axis and left y-axis) and stepwise heats (blue solid cycle, using top x-axis and right y-axis); b stepwise heat (right y axis, blue solid cycle—experimental Q, blue dashed line—fitted Q) and speciation of the O-phospho-l-serine (solid line, left y axis) versus the titrant volume. Initial solutions in the cup: V = 750 µL, \( {n_{{{\text{L}}^{3 - }}}} \) = 4.529 µmol, n +H = 13.587 µmol, titrant: 49.32 mmol·L−1 NaOH, injection volume: 5.0 μL
The stepwise enthalpies of protonation are − 38.7 ± 0.4 kJ·mol−1 (for H+ + L3− ⇋ HL2−), 0.1 ± 0.6 kJ·mol−1 (for H+ + HL2− ⇋ H2L−), and − 1.2 ± 1 kJ·mol−1 (for H+ + H2L− ⇋ H3L). It should be noted that the exact values of the enthalpies of protonation reaction for the second and third steps are much smaller than for the first step. The significant differences among the stepwise enthalpies for the protonation reaction suggests different environments for the hydrogen bond to O-phospho-l-serine. In the zwitterionic crystal form of O-phospho-l-serine, the N–H bond lengths (0.955, 0.833 and 0.794 Å) in the amino group (NH3 +) are much longer than O–H bond lengths in the phosphono group (0.766 Å) and carboxyl group (0.690 Å) [39]. This trend is in good agreement with the enthalpy of protonation directly determined by calorimetry. These results further confirm that the first protonation reaction occurs on the amine groups while the second and third protonation reactions occur on the oxygen atoms of the phosphonate group and carboxylate group.
4 Summary
The protonation of O-phospho-l-serine was evaluated by potentiometry, calorimetry, NMR spectroscopy and quantum chemical calculations. The overall protonation reactions are exothermic. Stepwise, the protonation reaction of fully deprotonated O-phospho-l-serine starts with the nitrogen atom of the amino group, followed by one of the oxygen atoms in the phosphono group, and finally the carboxyl oxygen atom. The protonation reaction occurring on the phosphono and carboxylate oxygen atoms are much smaller than on the nitrogen atom. The energetics of the protonation can be well explained by the difference in the nature of N–H and O–H bonds. The data from this work will help to understand possible speciation of O-phospho-l-serine in physiological and ecotoxicological processes in biochemical solutions.
References
Jastrzab, R., Lomozik, L.: Stability and coordination mode of complexes of polyphosphates and polymetaphosphates with copper(II) ions in aqueous solution—potentiometric, spectral and theoretical studies. J. Solution Chem. 39, 909–919 (2010)
Cohen, P.: The origins of protein phosphorylation. Nat. Cell Biol. 4, E127–E130 (2002)
Rogerson, D.T., Sachdeva, A., Wang, K., Haq, T., Kazlauskaite, A., Hancock, S.M., Huguenin-Dezot, N., Muqit, M.M.K., Fry, A.M., Bayliss, R., Chin, J.W.: Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat. Chem. Biol. 11, 496–503 (2015)
Kiss, E., Lakatos, A., Bányai, I., Kiss, T.: Interactions of Al(III) with phosphorylated amino acids. J. Inorg. Biochem. 69, 145–151 (1998)
Hendrickson, H.S., Fullington, J.G.: Stabilities of metal complexes of phospholipids: Ca(II), Mg(II), and Ni(II) complexes of phosphatidylserine and triphosphoinositide. Biochemistry 4, 1599–1605 (1965)
Jastrzab, R., Lomozik, L.: Coordination mode in the binary systems of copper(II)/O-phospho-l-serine. J. Coord. Chem. 62, 710–720 (2009)
Mohan, M.S., Abbott, E.H.: Metal complexes of amino acid phosphate esters. Inorg. Chem. 17, 2203–2207 (1978)
Mohan, M.S., Abbott, E.H.: Catalytic dephosphorylation of O-phosphoserine by gyloxylate ion and copper(II). Inorg. Chem. 17, 3083–3086 (1978)
Zachariou, M., Traverso, I., Spiccia, L., Hearn, M.T.W.: Potentiometric investigations into the acid–base and metal ion binding properties of immobilized metal ion affinity chromatographic (imac) adsorbents. J. Phys. Chem. 100, 12680–12690 (1996)
Fölsch, G., Österberg, R.: The apparent acid ionization constants of some O-phosphorylated peptides and related compounds. J. Biol. Chem. 234, 2298–2303 (1959)
Osterberg, R.: Metal and hydrogen-ion binding properties of O-phosphoserine. Nature 179, 476–477 (1957)
Homeyer, N., Horn, A.H.C., Lanig, H., Sticht, H.: Amber force-field parameters for phosphorylated amino acids in different protonation states: phosphoserine, phosphothreonine, phosphotyrosine, and phosphohistidine. J. Mol. Model. 12, 281–289 (2006)
Gran, G.: Determination of the equivalence point in potentiometric titrations. Part II. Analyst 77, 661–671 (1952)
Rossotti, F.J.C., Rossotti, H.: Potentiometric titrations using Gran plots: a textbook omission. J. Chem. Educ. 42, 375 (1965)
Gans, P., Sabatini, A., Vacca, A.: Investigation of equilibria in solution. determination of equilibrium constants with the hyperquad suite of programs. Talanta 43, 1739–1753 (1996)
Li, X., Zhang, Z., Endrizzi, F., Martin, L.R., Luo, S., Rao, L.: Effect of temperature on the protonation of N-(2-hydroxyethyl)ethylenediamine-N, N′, N′-triacetic acid in aqueous solutions: potentiometric and calorimetric studies. J. Chem. Thermodyn. 85, 35–41 (2015)
Liu, B., Dong, L., Yu, Q., Li, X., Wu, F., Tan, Z., Luo, S.: Thermodynamic study on the protonation reactions of glyphosate in aqueous solution: potentiometry, calorimetry and NMR spectroscopy. J. Phys. Chem. B. 120, 2132–2137 (2016)
Gans, P., Sabatini, A., Vacca, A.: Simultaneous calculation of equilibrium constants and standard formation enthalpies from calorimetric data for systems with multiple equilibria in solution. J. Solution Chem. 37, 467–476 (2008)
Hohenberg, P., Kohn, W.: Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964)
Kohn, W., Sham, L.J.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965)
Chai, J.-D., Head-Gordon, M.: Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008)
Ribeiro, R.F., Marenich, A.V., Cramer, C.J., Truhlar, D.G.: Use of solution-phase vibrational frequencies in continuum models for the free energy of solvation. J. Phys. Chem. B. 115, 14556–14562 (2011)
Marenich, A.V., Cramer, C.J., Truhlar, D.G.: Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 113, 6378–6396 (2009)
Wiberg, K.B.: Application of the Pople–Santry–Segal cndo method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24, 1083–1096 (1968)
Foster, J.P., Weinhold, F.: Natural hybrid orbitals. J. Am. Chem. Soc. 102, 7211–7218 (1980)
Reed, A.E., Weinstock, R.B., Weinhold, F.: Natural population analysis. J. Chem. Phys. 83, 735–746 (1985)
Reed, A.E., Curtiss, L.A., Weinhold, F.: Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem. Rev. 88, 899–926 (1988)
Moyano, A., Pericas, M.A., Valenti, E.: A theoretical study on the mechanism of the thermal and the acid-catalyzed decarboxylation of 2-oxetanones (β-lactones). J. Org. Chem. 54, 573–582 (1989)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery Jr., J.A., Peralta, J.E., Ogliaro, F., Bearpark, M.J., Heyd, J., Brothers, E.N., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A.P., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, N.J., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, Ö., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian, 09th edn. Gaussian Inc, Wallingford (2009)
Endrizzi, F., Melchior, A., Tolazzi, M., Rao, L.: Complexation of uranium(VI) with glutarimidoxioxime: thermodynamic and computational studies. Dalton Trans. 44, 13835–13844 (2015)
Alderighi, L., Gans, P., Ienco, A., Peters, D., Sabatini, A., Vacca, A.: Hyperquad simulation and speciation (Hyss): a utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 184, 311–318 (1999)
Jastrzab, R.: Studies of new phosphothreonine complexes formed in binary and ternary systems including biogenic amines and copper(II). J. Coord. Chem. 66, 98–113 (2013)
Jastrzab, R.: The influence of copper(II) ions on noncovalent interactions in the systems including phosphoserine and biogenic amines. New J. Chem. 34, 2867–2875 (2010)
Szpoganicz, B., Martell, A.E.: Thermodynamic and microscopic equilibrium constants of molecular species formed from pyridoxal 5′-phosphate and 2-amino-3-phosphonopropionic acid in aqueous and water-d2 solution. J. Am. Chem. Soc. 106, 5513–5521 (1984)
Gardiennet-Doucet, C., Assfeld, X., Henry, B., Tekely, P.: Revealing successive steps of deprotonation of l-phosphoserine through 13C and 31P chemical shielding tensor fingerprints. J. Phys. Chem. A 110, 9137–9144 (2006)
Potrzebowski, M.J., Assfeld, X., Ganicz, K., Olejniczak, S., Cartier, A., Gardiennet, C., Tekely, P.: An experimental and theoretical study of the 13C and 31P chemical shielding tensors in solid O-phosphorylated amino acids. J. Am. Chem. Soc. 125, 4223–4232 (2003)
McCallum, G.H., Robertson, J.M., Sim, G.A.: Crystal structure of l-serine phosphate. Nature 184, 1863–1864 (1959)
Bryndal, I., Picur, B., Lis, T.: Three polymorphic forms of dipotassium O-phospho-l-serinate hydrates. Z. Kristallog. 219, 38–46 (2004)
Sundaralingam, M., Putkey, F.F.: Molecular structures of amino acids and peptides. II. a redetermination of the crystal structure of l-O-serine phosphate. A very short phosphate-carboxyl hydrogen bond. Acta Cryst. B 26, 790–800 (1970)
Putkey, E.F., Sundaralingam, M.: Molecular structures of amino acids and peptides. I. the crystal structure and conformation of DL-O-serine phosphate monohydrate. Very short phosphate–phosphate hydrogen bonds. Acta Cryst. B 26, 782–789 (1970)
Maniukiewicz, W., Kwiatkowski, W., Blessing, R.H.: O-phospho-DL-threonine and O-phospho-l-threonine compared with their serine analogs. Acta Cryst. C. 52, 1736–1741 (1996)
Xie, Y., Jiang, Y., Ben-Amotz, D.: Detection of amino acid and peptide phosphate protonation using Raman spectroscopy. Anal. Biochem. 343, 223–230 (2005)
Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. 41573122) and China Academy of Engineering Physics for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liu, B., Tian, Y., Yu, Q. et al. Determination of Protonation Constants of O-Phospho-l-serine in Aqueous Solution: Potentiometry, Microcalorimetry, NMR Spectroscopy and Quantum Chemical Calculations. J Solution Chem 46, 2281–2292 (2017). https://doi.org/10.1007/s10953-017-0696-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-017-0696-6