
Abstract
N-(phosphonomethyl)-l-proline is an analogue of glyphosate. The protonation for N-(phosphonomethyl)-l-proline was studied by potentiometry, calorimetry, 31P NMR spectroscopy and quantum chemical calculations to further understand the protonation process of glyphosate. The results confirmed that the order of successive protonation sites of totally deprotonated N-(phosphonomethyl)-l-proline are a phosphonate oxygen, amino nitrogen, and finally the carboxylate oxygen. The results can improve the understanding of the biological activity of these types of molecules in solution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Protonation reactions are very important for a biological active molecule because of its ability to switch from a proton-acceptor to a proton-donor molecule with only small changes in the pH. This enables, for example, transporting protons from one molecule to another without wasting much energy on either binding or releasing the proton, which would inevitably be the case in a strictly proton-accepting or proton-donating molecule.
N-(phosphonomethyl)-l-proline (H3L) is an analogue of glyphosate with biological activity [1]. It appears that this molecule may be a potential substrate or inhibitor of the enzyme, which uses proline as its reagent. Proline and its analogs or homologs are very important chiral sources that have wide applications, for instance in stereo-selective synthesis, enantio separation and biochemistry. Due to its ability to coordinate in various fashions under different reaction conditions, N-(phosphonomethyl)-l-proline has been used to construct metal carboxylate-phosphonates with novel architectures [2,3,4,5,6,7,8]. However, the protonation of N-(phosphonomethyl)-l-proline in aqueous solution has not been reported so far. Thermodynamic data that describe the solution behavior of this molecule are scarce.
Both glyphosate and N-(phosphonomethyl)-l-proline molecules possess three donor groups, an amine group, a carboxylate group, and a phosphate group. The protonation reactions of N-(phosphonomethyl)-l-proline were investigated in this work by potentiometry, calorimetry and NMR spectroscopy. The results from this similar molecule will provide further evidence concerning the protonation steps of glyphosate and be helpful in gaining further insights into the chemical processes of glyphosate in the environment.
2 Experimental
2.1 Chemicals
All chemicals were reagent-grade or higher quality. Milli-Q water was boiled to remove carbon dioxide and then used for the preparation of all solutions. The chemicals used in the synthesis of N-(phosphonomethyl)-l-proline (H3L) were all purchased from Aladdin (China). The ligand was prepared according to the method reported in the literature [9, 10]. The purity, checked by alkalimetric titration, was higher than 99%. A stock solution of the ligand was prepared by dissolving the desired amount of the solid in a solution containing 0.5 mol·L−1 NaCl (≥98%, Aladdin). Working solutions of NaOH and HCl containing NaCl were standardized by titration with potassium hydrogen phthalate (primary standard, Alfa Aesar) and Trizma base (crystalline, Sigma), respectively. Carbonate contamination in NaOH titrant solution is less than 0.5%, determined from blank acid–base titrations by Gran’s method [11]. The ionic strength of all working solutions was maintained at 0.5 mol·L−1 (NaCl). Deuterium oxide (99.8% D, J&K) was used to prepare sample solutions for NMR experiments.
2.2 Potentiometry
The electromotive force (emf, in millivolts) was measured using a potentiometric titrator (888 Titrando, Metrohm) equipped with a combination pH electrode (6.0259.100 Unitrode, Metrohm), in an argon atmosphere to avoid the interference of carbon dioxide. All titrations were carried in a 100 mL capped cell. Both the cap and the cell were water-jacketed and maintained at 25.0 °C by circulating water from a constant-temperature bath [12, 13]. The uncertainty of temperature is ±0.1 °C. Prior to each titration, an acid–base titration with standard HCl and NaOH solution was performed to obtain the electrode parameters, which allowed the calculation of hydrogen ion concentrations from the electrode potential in the subsequent titrations, conducted at the same condition of temperature and ionic strength. A detailed description of the calibration of electrodes has been provided elsewhere [13]. To minimize systematic errors and to check the repeatability of the measurements, multiple titrations were conducted with solutions containing different concentration of the ligand (C L as total ligand concentration) and acidity (C H for total hydrogen ion concentration). Usually, about 50 points were collected for each titration. The protonation constants (log10 β) were calculated by a nonlinear regression program [14].
2.3 Microcalorimetry
Microcalorimetric titrations at 25.0 ± 0.1 °C were performed on an isothermal titration microcalorimeter (TAM-III) that measures the heat flow among the reaction vessel, reference vessel, and a heat sink maintained at a constant temperature [12, 13]. Both chemical and the electrical calibrations were performed to validate the performance of the instrument. The ligand was maintained in the 750 μL reaction vessel and stirred with a gold propeller at 80 rpm. The titrant (~50 mmol·L−1 NaOH) was added into the vessel through a Hamilton 250 μL syringe with stepwise 5 μL additions. About 50 injections were made in each experiment. Multiple titrations using different ligand and acid concentrations were performed to reduce the uncertainty of the results. The observed reaction heat (“partial” or stepwise Q) is a function of a number of parameters, including the concentrations of reactants, the protonation constants (log10 β) and the enthalpy of protonation of the ligand (ΔH). Using the stoichiometric concentrations of the reactants and the protonation constants measured by potentiometry in this work, the enthalpies for protonation of N-(phosphonomethyl)-l-proline at 25 °C were calculated from the calorimetric titration data [15].
2.4 NMR Spectroscopy
31P NMR experiments were performed on a 400 MHz Bruker Avance spectrometer. Samples were prepared by dissolving 80 mg solid N-(phosphonomethyl)-l-proline in 4 mL of D2O. After the ligand was completely dissolved, the whole solution was divided equally into four parts. The pH of each part solution was adjusted by adding HCl or NaOH to cover a wide pH region (1.5–13.5). An aliquot of each solution was pipetted into an NMR sample tube for the NMR experiments. H3PO4 in D2O was used as reference and the shift of 31P for H3PO4 in D2O was set as 0 ppm. The protonation constants for H2O solutions were assumed to be applicable. The acidities (expressed as pD = −log10[D+]) in D2O were obtained by using pD = pH + 0.40 [16].
2.5 Quantum Chemical Calculations
The electron correlation effects were included by employing density functional theory (DFT) [17, 18] methods at the PBE0-D3 (BJ) [19,20,21,22] level of theory. The triple-ζ basis set 6–311 + G(d, p) was used to describe the H, C, N, O and P atoms. The default fine grid (75, 302), having 75 radial shells and 302 angular points per shell, was used to evaluate the numerical integration accuracy. All of the species were optimized in aqueous solution while using the conductor-like polarized continuum model (CPCM) [23, 24] model with universal force field (UFF) radii [25, 26]. The natural atomic charges and Wiberg bond indices (WBIs) [27] were calculated on the basis of natural bond orbital (NBO) [28,29,30] theory. The WBIs between two atoms are a measure of the bond order and, hence, of the bond strength between these two atoms [31]. All calculations were carried out with the Gaussian 09 program [32].
3 Results and Discussion
3.1 Protonation Constants of N-(phosphonomethyl)-l-proline
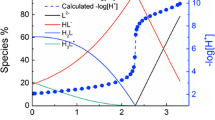
Representative potentiometric titrations of N-(phosphonomethyl)-l-proline at 25 °C and the fitting curves are shown in Fig. 1. There are three successive protonation reactions in the pH range of this study. The fourth protonation reaction, on the second oxygen atom of the phosphonate group, occurs outside the normal pH range and so is not considered here. The speciation during the course of the titration experiment is superimposed on these plots. The protonation constants (log10 β) of the molecule were calculated and are reported in Table 1. The protonation constants of glyphosate from the literature are also listed in Table 1 for comparison. The protonation constants of N-(phosphonomethyl)-l-proline are larger than those of glyphosate by one order of magnitude. The trend reflects the electron density on the central amine: the electron-donating pyrrolidine ring makes N-(phosphonomethyl)-l-proline more basic than glyphosate.

Potentiometric titrations for the protonation of N-(phosphonomethyl)-l-proline at 25 °C; I = 0.5 mol·L−1 NaCl. Initial solution: V = 20.95 mL, \( n_{{{\text{L}}^{3 - } }} = 0.1120\;{\text{mmol}} \), \( n_{{{\text{H}}^{ + } }} = 0. 3 9 4 9 {\text{ mmol}} \), titrant: 73.20 mmol·L−1 NaOH. Symbols: red solid square, experimental data (−log10[H+]); black dashed line, fitting (−log10[H+]); solid lines, percentages of species relative to the total N-(phosphonomethyl)-l-proline concentration (black, H3L; red, H2L−; blue, HL2−; green, L3−, where H3L stands for the neutral N-(phosphonomethyl)-l-proline in solution) (Color figure online)
3.2 Enthalpy of Protonation of N-(phosphonomethyl)-l-proline
Calorimetric information provides revealing insights into solute–solvent interactions. This improved understanding of solute behavior is particularly useful in studying complex biomolecules such as proteins, where an accurate thermodynamic description of the component amino acids may help to elucidate the structure, folding, and other functions of biomolecules. Data of the calorimetric titrations for the protonation of N-(phosphonomethyl)-l-proline are shown in Fig. 2. The calculated enthalpies for protonation of the ligand at 25 °C are also presented in Table 1. Stepwise, the enthalpies of protonation reaction are −33.0 ± 0.3 kJ·mol−1 (for H+ + L3−⇌ HL2−), −0.1 ± 1 kJ·mol−1 (for H+ + HL2− ⇌ H2L−), and −16.9 ± 3.0 kJ·mol−1 (for H+ + H2L−⇌ H3L). It should be pointed out that the value of the enthalpy of the second protonation reaction is much smaller than for the first and third steps. It was well known that amine nitrogen atoms are less hydrated than the oxygen atoms of carboxylate or phosphate groups in aqueous solution. So, less energy is released upon amino nitrogen protonation compared to protonation of the carboxyl or phosphono groups. For example, glutaroimide-dioxime molecule (H2A) is a cyclic imidedioxime moiety that can form during the synthesis of the poly-amidoxime sorbent and is reputedly used for the extraction of uranium from seawater [33]. There are three steps of protonation, from A2−, through HA− and H2A, and then to H3A+. The acid strengths of the oxime groups are very weak. The first two stepwise protonated sites are oxygen atoms of the oxime group and the third protonation reaction occurs on the nitrogen atom [34]. The first two stepwise enthalpies for the protonation of glutarimidedioxime are −36.1 kJ·mol−1 (for H+ + A2−⇌ HA−) and −33.6 kJ·mol−1 (for H+ + HA−⇌ H2A), while the enthalpy change for last step protonation reaction is 7.3 kJ·mol−1 (for H+ + H2A ⇌ H3A+) [33]. Another example is that aminocarboxylic acids, which exist in aqueous solutions as zwitter ions, where the amine nitrogen is protonated and possesses a positive charge, leaving the carboxylate oxygen unprotonated with a negative charge. The variations of enthalpies for the protonation of N-(phosphonomethyl)-l-proline suggest that the second protonation occurs on the amino nitrogen atom while the first and third protonations occur on oxygen atoms.

Calorimetric titrations for the protonation of N-(phosphonomethyl)-l-proline at 25 °C, I = 0.5 mol·L−1 NaCl. a Thermogram (solid line, using bottom x-axis and left y-axis) and stepwise heat (navy point, using top x-axis and right y-axis). b Stepwise heat (right y axis, navy point-experimental Q, black dashed line-fitted Q) and speciation of N-(phosphonomethyl)-l-proline (solid line, left y axis, pink: H3L, blue: H2L−, red: HL2−, green: L3−, where H3L stands for the neutral N-(phosphonomethyl)-l-proline molecule in solution) versus the titrant volume. Initial solutions in cup: V = 750 µL, \( n_{{{\text{L}}^{3 - } }} = 3.0 6 5 { }\mu {\text{mol}} \), \( n_{{{\text{H}}^{ + } }} = 1 3. 2 3 9 { }\mu {\text{mol}} \). Titrant: 49.92 mmol·L−1 NaOH, injection volume: 5.0 μL (Color figure online)
3.3 31P NMR
NMR spectroscopy is a powerful technique for studying both the qualitative and quantitative relations among different organic molecules [35–37]. For example, NMR spectroscopy can provide reliable pK a data below pH = 2 and above pH = 12 when potentiometric titration can not accurately discriminate between water’s reactions with the base or acid and those of the ligand [38]. 31P NMR spectra of four solutions (solutions a, b, c, and d) were collected to help identify the protonation sites of the molecule (Fig. 3). The acidities (expressed as pD = −log10[D+]) of the solutions are 13.1, 7.1, 3.4, and 1.5, for solutions a, b, c, and d, respectively. The dominant species of N-phosphonomethy-l-proline in these solutions are calculated to be as following: solution a, L3− (100%); solution b, HL2− (98%); solution c, H2L− (97%), and solution d, H3L (90%). Therefore, the variations in 31P NMR spectra from solution a to d can be discussed as reflecting the stepwise protonation of totally deprotoned N-(phosphonomethyl))-l-proline.

31P spectra of four N-(phosphonomethyl)-l-proline solutions. The acidities (expressed as pD = −log10[D+]) of the solutions are: a 13.1, b 7.1, c 3.4, and d 1.5, respectively
Usually, as the protonation reaction occurs closer to the phosphorus atom, the larger is the 31P NMR spectra chemical shift (i.e., the ‘‘proximity effect’’) [12, 39]. As shown in Fig. 3, in going from solution a to b, the dominant species changes from L3− to HL2−, representing the first protonation step (H+ + L3−⇌ HL2−). The change in the chemical shift (~7.49 ppm) is the largest, suggesting the first protonation occurs on the phosphonate oxygen atom as described by Fig. 4a, b. From solution b to solution c, the dominant species changes from HL2− to H2L−, representing the second protonation step (H+ + HL2−⇌ H2L−). The chemical shift change (∼1.50 ppm, Fig. 3b, c) is smaller than the first protonation step, but larger than that (∼0.37 ppm, Fig. 3c, d) of the third protonation step (H+ + H2L− = H3L), which suggests that the second protonation occurs on the amino nitrogen atom and the third protonation occurs on the carboxylate oxygen atom. Generally, the basicities of the amino and imino nitrogen atoms are higher than that of a carboxyl group. The successive protonation sites of totally deprotonated N-(phosphonomethyl)-l-proline in order are expressed as Fig. 4.

Structures of N-(phosphonomethyl)-l-proline connected with consecutive protonation reactions
3.4 Conformational Study of the HL2− Species of N-(phosphonomethyl)-l-proline
To gain deeper insights into this protonation reaction, we further studied this process by quantum chemical methods. The best solvation model should involve the explicit inclusion of waters in the first hydration shell combined with a continuum solvation model for the remainder of the hydration shell [40]. Likewise, for comparison, the continuum solvation model was also used for all of the hydration shell in this work.
The structure of three possible protonation sites on N-(phosphonomethyl)-l-proline for the first protonation species (HL2−): the phosphonate (OP), the amine (N), and the carboxyl (OC), were evaluated by the DFT calculations in aqueous medium. The optimized structures of the stationary points are described in Fig. 5a, b and c. As shown in Fig. 5d, the order of the relative energies for the three situations is H–L(OP) < H–L(N) < H–L(OC), suggesting that the first protonation reaction at the phosphonate group forms the most stable structure. As depicted in Table 2, moreover, the magnitude of WBIs is H–L(OP) > H–L(N) > H–L(OC), which also is in good accordance with the changes of energies. These calculations indicate that the first protonation reaction should occur upon one of oxygen atoms of the phosphonate group.

The optimized structures for the first protonation speciation (HL2−) by DFT calculations: a HL2−, protonation on phosphonate; b HL2−, protonation on amine; c HL2−, protonation on carboxyl (the H–L bond lengths are also shown in the figure); d the relative energies for various possible HL2− speciations; (d-1) the continuum solvation model for all of the hydration shell; (d-2) the explicit inclusion for the first hydration shell and continuum solvation model for the remainder of the hydration shell which corresponds to three optimized structures in (a), (b) and (c), respectively
3.5 Implication to Environmental Solutions
Most herbicides have acidic or basic functionalities. Depending on the both pH and the protonation constants of active ingredient in the herbicide, different chemical species may exist in the solution, including cationic, anionic, or neutral species, which generally have different properties. Because both glyphosate and N-(phosphonomethyl)-l-proline molecules possess the same donor groups, the results from this study further prove the first protonation site of glyphosate is on the phosphonate group. The pH value is 7–8 for most biochemical solutions, surface waters, and ground waters. For these aqueous solutions, the dominant species of glyphosate and N-(phosphonomethyl)-l-proline is HL2−. Our results confirm that the dominant species of glyphosate and N-(phosphonomethyl)-l-proline molecules in pH range 7–8 do not have the proton on the amino group but rather on the phosphonate group (as shown in Fig. 6). We hope that, once the most stable glyphosate species are determined, this knowledge may help in the study of the corresponding complexation reactions of glyphosate with different metals found in the soil.

Dominant species of glyphosate and N-(phosphonomethyl)-l-proline in the pH range 7–8: a glyphosate, b N-(phosphonomethyl)-l-proline
4 Summary
Protonation reactions of an analogue of glyphosate, N-(phosphonomethyl)-l-proline, has been studied by thermodynamic experiments and quantum chemical calculations. The results prove that the protonation reactions of fully deprotonated N-(phosphonomethyl)-l-proline start with one of the oxygen atoms of the phosphonic group, followed by the amine group’s nitrogen atom, and finally the carboxyl oxygen atom. This result further proves that the first protonation site of glyphosate is not on the amino group but on the phosphonate group instead. The data from this work will help to understand glyphosate and N-(phosphonomethyl)-l-proline chemical actions in biochemical solutions, surface waters, and ground waters.
References
Sawka-Dobrowolska, W., Barycki, J.: Structure of N-phosphonomethyl-l-proline. Acta Crystallogr. C 45(4), 606–609 (1989)
Li, H., Liu, Y., Huo, Q.: Hydrothermal synthesis, crystal structure and magnetic properties of a novel nickel carboxylate–phosphonate with a layered structure. Inorg. Chem. Commun. 38, 33–38 (2013)
Shi, X., Zhu, G., Qiu, S., Huang, K., Yu, J., Xu, R.: Zn2[(S)-O3PCH2NHC4H7CO2]2: a homochiral 3D zinc phosphonate with helical channels. Angew. Chem. Int. Ed. 43(47), 6482–6485 (2004)
Huang, K., Yu, J., Wang, G., Li, Y., Xu, R.: Covalent bonding of phosphonates of l-proline and l-cysteine to γ-zirconium phosphate. Eur. J. Inorg. Chem. 2004(14), 2956–2960 (2004)
Turner, A., Jaffres, P.-A., MacLean, E.J., Villemin, D., McKee, V., Hix, G.B.: Hydrothermal synthesis and crystal structure of two Co phosphonates containing trifunctional phosphonate anions: Co3(O3PCH2NH2CH2PO3)2 and Co3(O3PCH2-NC4H7-CO2)2 5H2O. Dalton Trans. 7, 1314–1319 (2003)
Yang, B.-P., Mao, J.-G., Sun, Y.-Q., Zhao, H.-H., Clearfield, A.: Syntheses, characterizations, and crystal structures of three new metal phosphonocarboxylates with a layered and a microporous structure. Eur. J. Inorg. Chem. 2003(23), 4211–4217 (2003)
Sun, Z.-G., Cui, L.-Y., Liu, Z.-M., Dong, D.-P., Meng, L., Chen, H., Zhang, L.-C., Zhu, Z.-M., You, W.-S.: Hydrothermal synthesis and crystal structure of a novel lead(II) phosphonate containing trifunctional phosphonate anions: Pb4O[O3PCH2–NC4H7–CO2]2. Inorg. Chem. Commun. 9(11), 1121–1124 (2006)
Yang, B.-P., Mao, J.-G.: Homochiral cobalt(II) and strontium(II) amino-carboxylate–phosphonate hybrids. J. Mol. Struct. 830(1–3), 78–84 (2007)
Yue, Q., Yang, J., Li, G.-H., Li, G.-D., Chen, J.-S.: Homochiral porous lanthanide phosphonates with 1D triple-strand helical chains: synthesis, photoluminescence, and adsorption properties. Inorg. Chem. 45(11), 4431–4439 (2006)
Diel, P.J., Maier, L.: Organische Phosphorverbidndungen 78 Hertellung und Eigenschaften von N-phosonomethylglycin Derivaten. Phosphorus Sulfur Relat. Elem. 20(3), 313–321 (1984)
Gran, G.: Determination of the equivalence point in potentiometric titrations. Part II. Analyst 77(920), 661–671 (1952)
Liu, B., Dong, L., Yu, Q., Li, X., Wu, F., Tan, Z., Luo, S.: Thermodynamic study on the protonation reactions of glyphosate in aqueous solution: potentiometry, calorimetry and NMR spectroscopy. J. Phys. Chem. B 120(9), 2132–2137 (2016)
Li, X., Zhang, Z., Endrizzi, F., Martin, L.R., Luo, S., Rao, L.: Effect of temperature on the protonation of N-(2-hydroxyethyl)ethylenediamine-N, N′, N′-triacetic acid in aqueous solutions: potentiometric and calorimetric studies. J. Chem. Thermodyn. 85, 35–41 (2015)
Gans, P., Sabatini, A., Vacca, A.: Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 43(10), 1739–1753 (1996)
Gans, P., Sabatini, A., Vacca, A.: Simultaneous calculation of equilibrium constants and standard formation enthalpies from calorimetric data for systems with multiple Equilibria in solution. J. Solution Chem. 37(4), 467–476 (2008)
Glasoe, P.K., Long, F.A.: Use of glass electrodes to measure acidities in deuterium oxide. J. Phys. Chem. 64(1), 188–190 (1960)
Hohenberg, P., Kohn, W.: Inhomogeneous electron gas. Phys. Rev. 136(3B), B864–B871 (1964)
Kohn, W., Sham, L.J.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140(4A), A1133–A1138 (1965)
Grimme, S., Antony, J., Ehrlich, S., Krieg, H.: A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements HPu. J. Chem. Phys. 132(15), 154104 (2010)
Grimme, S., Ehrlich, S., Goerigk, L.: Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32(7), 1456–1465 (2011)
Goerigk, L., Reimers, J.R.: Efficient methods for the quantum chemical treatment of protein structures: the effects of London-dispersion and basis-set incompleteness on peptide and water-cluster geometries. J. Chem. Theory Comput. 9(7), 3240–3251 (2013)
Adamo, C., Barone, V.: Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110(3), 6158–6170 (1999)
Barone, V., Cossi, M.: Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 102(11), 1995–2001 (1998)
Cossi, M., Rega, N., Scalmani, G., Barone, V.: Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24(6), 669–681 (2003)
Rayne, S., Forest, K.: Theoretical studies on the pKa values of perfluoroalkyl carboxylic acids. J. Mol. Struct. Theochem 949(1–3), 60–69 (2010)
Marque, S., Razafimahaleo, V., Dinut, A., Grach, G., Prim, D., Moreau, X., Gil, R.: On the molecular structure and geometry of pyridylalkylamine-H+ complexes: application to catalytic enantioselective hydroxyalkylation of indoles. New J. Chem. 37(9), 2683–2690 (2013)
Wiberg, K.B.: Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24(3), 1083–1096 (1968)
Foster, J.P., Weinhold, F.: Natural hybrid orbitals. J. Am. Chem. Soc. 102(24), 7211–7218 (1980)
Reed, A.E., Weinstock, R.B., Weinhold, F.: Natural population analysis. J. Chem. Phys. 83(2), 735–746 (1985)
Reed, A.E., Curtiss, L.A., Weinhold, F.: Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88(6), 899–926 (1988)
Moyano, A., Pericas, M.A., Valenti, E.: A theoretical study on the mechanism of the thermal and the acid-catalyzed decarboxylation of 2-oxetanones (beta-lactones). J. Org. Chem. 54(3), 573–582 (1989)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, E.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian 09 ed. Gaussian, Inc., Wallingford CT (2009)
Tian, G., Teat, S.J., Zhang, Z., Rao, L.: Sequestering uranium from seawater: binding strength and modes of uranyl complexes with glutarimidedioxime. Dalton Trans. 41(38), 11579–11586 (2012)
Mehio, N., Williamson, B., Oyola, Y., Mayes, R.T., Janke, C., Brown, S., Dai, S.: Acidity of the poly(acrylamidoxime) adsorbent in aqueous solution: determination of the proton affinity distribution via potentiometric titrations. Ind. Eng. Chem. Res. 55(15), 4217–4223 (2016)
Zhang, Z., Gibson, P., Clark, S.B., Tian, G., Zanonato, P.L., Rao, L.: Lactonization and protonation of gluconic acid: a thermodynamic and kinetic study by potentiometry, NMR and ESI-MS. J. Solution Chem. 36(10), 1187–1200 (2007)
Chruszcz, K., Barańska, M., Czarniecki, K., Boduszek, B., Proniewicz, L.M.: Experimental and calculated 1H, 13C and 31P NMR spectra of pyridine-2-phosphono-4-carboxylic acid. J. Mol. Struct. 648(3), 215–224 (2003)
Hummel, M., Leppikallio, M., Heikkinen, S., Niemelä, K., Sixta, H.: Acidity and lactonization of xylonic acid: a nuclear magnetic resonance study. J. Carbohydr. Chem. 29(8–9), 416–428 (2010)
Mehio, N., Lashely, M.A., Nugent, J.W., Tucker, L., Correia, B., Do-Thanh, C.L., Dai, S., Hancock, R.D., Bryantsev, V.S.: Acidity of the amidoxime functional group in aqueous solution: a combined experimental and computational study. J. Phys. Chem. B 119(8), 3567–3576 (2015)
Maki, H., Ueda, Y., Nariai, H.: Protonation equilibria and stepwise hydrolysis behavior of a series of thiomonophosphate anions. J. Phys. Chem. B 115(13), 3571–3577 (2011)
Shamov, G.A., Schreckenbach, G.: Density functional studies of actinyl aquo complexes studied using small-core effective core potentials and a scalar four-component relativistic method. J. Phys. Chem. A 109(48), 10961–10974 (2005)
Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. 41573122). Calculations were done on the computational grids in the Supercomputing Center of Chinese Academy of Sciences (SCCAS).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Li, Q., Liu, B., Tian, Y. et al. Thermodynamics of the Protonation of an Analogue of Glyphosate, N-(phosphonomethyl)-l-proline, in Aqueous Solutions. J Solution Chem 46, 1048–1058 (2017). https://doi.org/10.1007/s10953-017-0620-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-017-0620-0