
Abstract
The acid–base properties of 2-thiobarbituric, 1,3-diethyl-, and 1,3-dibutyl-2-thiobarbiturc acids and the thermodynamics of their complexation with Ag(I) ions in aqueous solutions have been investigated using a spectrophotometric technique and ab initio calculations. The free acids primarily exist in keto-form and undergo enolization in neutral aqueous solutions. They undergo protonation via free oxygen atoms in acidic conditions. It has been discovered that 1,3-diethyl-2-thiobarbiturc acid behaves as a S-donor ligand under these conditions. The TD–DFT calculations of all the thiobarbiturc acids have revealed π–π* transitions from the sulfur and oxygen atoms in the ring. The complexation with Ag(I) ions lead to the transition of the z2 orbital of silver and π orbitals of sulfur into π* orbitals of a ring.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Thiobarbituric acid (H2TBA) and its 1,3-derivatives demonstrate pronounced biological and coordinating properties, being used in various branches of medical science, pharmaceutical industry and chemical technology. Moreover, H2TBA being translated in reactive substances (TBARs) is widely used in biological studies [1–7] and research related to the food chemistry [8–10]. Some fluorine derivatives exhibit antiseptic and anti-diabetic activities [11] and can be used against non-alcoholic fatty liver disease [12, 13]. Like H2TBA, its 1,3-derivatives, like 1,3-diethyl-2-thiobarbituric acid (H2DETBA), can serve as an indicator of the oxidation of fats [14]. The individual properties of 1,3-dibutyl-2-thiobarbituric (H2DBTBA) acid have been investigated neither in solution nor in solid state.
Research dedicated to potentiometric investigations of complexation of H2TBA with lanthanides [15], actinides [16], and some transition and noble metals in aqueous solutions [17–19] have proposed flat S-coordinated structures for all species. These properties can be applied in analytical chemistry of some transition metals [19]. Moreover, some of them, for instance complexes of tin, exhibit biological activity [20]. The possibility of solid phase extraction of Ru(III) based on H2TBA polymers has been suggested [21]. It has been established that solid state complex species of H2DETBA with alkali metals and lead show O- and S-coordination [22, 23].
We may conclude that the investigations dedicated to the behavior of these ligands as biological reagents in aqueous solutions are of great interest. Unfortunately, the solution chemistry of barbituric and thiobarbituric acids is poorly investigated. Ag(I) has been chosen due to simplicity of complex formation of silver with other heterocyclic system [24]. Also, Ag(I) can be used as an antimicrobial agent in medical devices [25]. Here the Ag–thiobarbituric system serves as a model for further studies of complexation reactions of these ligands with other metals.
The current research deals with an integrated approach (spectrophotometric technique and DFT simulations) to a description of the fundamental equilibria in aqueous solutions. For this goal, acid–base and tautomeric properties of H2TBA, H2DETBA, and H2DBTBA, and their complexation processes with Ag(I) have been investigated.
2 Experimental Section
2.1 Chemicals Used
All chemicals were of analytical grade: 2-thiobarbituric acid (Aldrich ≥ 99 %), 1,3-diethyl-2-thiobarbituric acid (Aldrich ≥ 98 %), 1,3-dibutyl-2-thiobarbituric acid (AlfaAesar ≥ 97 %), HCl, H2SO4, HClO4, KCl, NH2CH2COOH, CH3COONa, CH3COOH, NaClO4, AgClO4. All chemicals were used as received.
All stock solutions were obtained by dissolution of dry salts and ligand weights. The metal salts and ligands were dissolved in distilled water. Concentration of mineral acid (HCl, H2SO4, and HClO4) was determined titrimetrically with standardized Na2CO3 solution.
Buffer solutions within the pH range from 1.00 to 2.20 were prepared with HCl and KCl (or HClO4 and NaClO4), from 2.20 to 3.60 pH with NH2CH2COOH and HCl, and from 3.60 to 5.60 pH with CH3COOH and CH3COONa. The desired pH values were obtained by adjusting the molarities of the buffer components [26]. The protonation equilibriums of thiobarbituric acids were investigated spectrophotometrically in hydrochloric, perchloric, and sulfuric acid aqueous solutions.
2.2 Apparatus and Procedure
The UV–Vis spectra were measured with an Evolution 300 scanning spectrophotometer (ThermoScientific, UK) using 1 cm quartz cells. Cell thermostating (±0.1 K) was performed with the Haake K15 thermostat connected to the Haake DC10 controller. The absorbances of solutions were measured between 220 and 450 nm. All measurements were performed at 298 K.
2.3 Ab Initio Study
Calculations were performed using the NWChem [27] program package on the Super computer of the Institute of Space and Information Technologies (SFU) [28]. Geometry optimization was performed by density functional theory (DFT) with the hybrid functional PBE0 [29]. The cc-pVDZ [30] basis set was applied for C, S, N, H and O atoms. The cc-pVDZ-PP [31] basis set including pseudopotential for Ag was applied. The solvent effects were evaluated using the SMD (COSMO) solvation model [32]. The UV–Vis spectra of complex species were reproduced from the vertical excitation energies for the first 11 singlet excited states (TD–DFT/PBE0/SMD). The optimized geometries were visualized with the ChemCraft software [33].
2.4 UV–Vis Measurements
Conditional stability constants (β) for monocomplex species were calculated from the equations:
where A λ is an absorbance at a given wavelength and C M and C H2L are the analytical concentrations of Ag(I) and H2DETBA, respectively.
The values of dissociation constants (pK a) have been calculated using the following equation [34]:
together with the Henderson–Hasselbach equation [35]:
where I is an ionization ratio.
The Cox–Yates method [36] based on the excess acidity function χ [37] was used to determine the protonation constant (K H) in strongly acidic solutions:
where A i , \( A_{{{\text{H}}_{2} {\text{L}}}} (\varepsilon_{{{\text{H}}_{2} {\text{L}}}} ) \), \( A_{{{\text{H}}_{3} {\text{L}}^{ + } }} (\varepsilon_{{{\text{H}}_{3} {\text{L}}^{ + } }} ) \) and \( A_{{{\text{HL}}^{ - } }} (\varepsilon_{{{\text{HL}}^{ - } }} ) \) are the absorbances and molar extinction coefficients of the process solution, the free ligands, and their conjugate acid or base, respectively [34, 38]. Calculation of all equilibrium constants and molar extinction coefficients was performed using Scilab 5.5 software [39] by means of nonlinear LSR analysis [40]:
3 Results and Discussion
The neutral ligand may exist in four tautomeric forms (Scheme 1). The isomers of H2TBA with protons at the 1 and 3 positions (Supplementary Figs. S1–S3) are energetically unfavorable [41, 42]. In solution the keto–enol equilibrium strongly depends on the solvent [43] and water stabilizes the enol form, however reliable experimental data on the equilibria of these ligands in solution are absent. To fill this gap the calculations of the keto–enol equilibrium (with inclusion of non-specific solvation) were performed to determine the reactive form.

Keto–enol and acid–base equilibrium (R = H, C2H5, C4H9; L = TBA, DETBA, DBTBA)
The keto-isomer is the most energetically favorable among the neutral forms existing in solution (Table 1). Protonation of all three neutral molecules leads to the formation of six species (Scheme 1, H3L+; Supplementary Figs. S4–S6), and the enols with proton bonded to the oxygen atom are the most energetically favorable species (Table 1). Supplementary Tables S1 and S2 show the absolute values of calculated Gibbs energies.
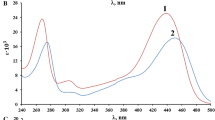
As can be seen from Fig. 1a–c), the spectra of neutral forms at C(HCl) = 1 mol·L−1 have two absorption maxima (in the case of H2DETBA and H2DBTBA one strong maximum and a shoulder). The numbers in parentheses specify the conditions under which the spectra were measured. The spectra of anions and cations have 3 and 1 absorption peaks, respectively. The theoretical EAS (Fig. 1a1–c1) calculated with TD–DFT for the most stable forms is shifted on 20–30 nm in the short-wave region of the experimental spectrum. The calculated EAS of anion species shows a considerable discrepancy with the experimental data (Fig. 1). The predominance of the keto–form in the aqueous solutions was indicated by agreement between the calculated profile and the experimental spectrum. The experimental and calculated spectra are similar in form and intensity ratios among the peaks. It is worth mentioning that these molecules are quite difficult for TD–DFT simulations.

UV–Vis spectra (a, b and c) and TD–DFT calculated (a1, b1 and c1) EAS. 1—Neutral (C(HCl) = 1 mol·L−1), 2—anion (pH = 5.6), 3—cation (C(HCl) > 9.5 mol·L−1) forms, 4—enol neutral form (calculated). Doppler broadening—20 band width on ½ height
Table 2 shows the values of molar extinction coefficients of the ligands at different wavelengths. The complication of radicals’ structure in 1,3-positions leads to a general decrease of the molar extinction coefficients in the region of 260–300 nm (Fig. 1). The position of the absorption maximum and value of the extinction coefficient in the region of 220–270 nm remain unchanged during dissociation (in pH region) for all acids.
However, changes can be observed in the region of 270–320 nm. On the contrary, dissociation and protonation processes (in strongly acidic solutions) change the absorption pattern significantly only at shorter wavelengths (220–270 nm). A linear relationship was found between absorbance and concentration (C(HCl) = 1 mol·L−1, C(H2L) = 10−3–10−5 mol·L−1) for all three ligands (indicating the absence of any dimers or ionic associates). Figure 2 show an example of the change in absorbance for dissociation (a), protonation (b) and complex formation (c). All raw spectroscopic data are given in the Supplementary (Supplementary Tables S3–S5; Supplementary Figs. S7–S10). Three isobestic points (Fig. 2a) indicate the presence of only two absorbing forms, a neutral molecule and an anionic species. The analysis of the log10 I–pH relationship has shown one proton being removed with increasing pH (Supplementary Fig. S11).

The UV–Vis spectra and absorbance at single wavelength: a H2DBTBA—pH (C(H2DBTBA) = 2.3 × 10−5 mol·L−1, I = 0.25 mol·L−1); b H2TBA—log10[C(HCl)], (C(H2TBA) = 1.36 × 10−4 mol·L−1); c H2DETBA—C(Ag) (C(H2DETBA) = 2.05 × 10−4 mol·L−1, C(H+) = 0.5 mol·L−1, I = 2 mol·L−1)
Figure 2b shows the H2TBA spectra in strong hydrochloric acidic media along with the A 235–log10[C(HCl)] relationship. Unfortunately, for the reasons described below, the experimental values of the molar extinction coefficients of fully protonated forms have not been determined. However, both the high concentrations of acid and the significant decrease in the absorbance testify to the dominance of the protonated form in solution.
Study of the acid–base properties of these ligands in sulfuric acid is impossible. In sulfuric acid, at any concentration, all ligands undergo restructuring [44, 45] with the formation of a yellow product (Supplementary Fig. S12). The investigation of silver complexation with H2TBA and H2DBTBA in aqueous solutions has been impossible due to the high insolubility of the products. The investigation of monocomplex species in the Ag–H2DETBA system has been performed under conditions of metal excess, for acidity 0.5, 1.0 and 1.5 mol·L−1 HClO4 with I = 2 mol·L−1. Figure 2c shows a typical UV–Vis spectrum and A 313–C Ag relationship for this system. Since the ∆A value at 310 (Supplementary Fig. S13) nm remains invariant at different Ag+ concentrations, one may conclude that the monocomplex AgH n DETBA is the only product. Also, the contribution from poly-nuclear Ag m H n DETBA species is practically negligible.
The values of acid–base equilibrium constants are given in Table 3. There are a number of potentiometric works dedicated to the determination of pK a(H2TBA) values showing quite different results. The pK a values range from 1.8 to 2.3 [46–48]. Spectrophotometric investigation has given similar results [46]. Table 3 shows thiobarbituric acid to be the strongest acid among the others. Its 1,3-derivatives have almost identical pK a values, which can be explained by the inductive effect of the alkyl groups. All the investigated acids are stronger than barbituric acid (pK a = 4.05; I = 0.5 [49]), but weaker than some arildene-thiobarbituric acids [50].
The values of protonation constants range from 2.2 to 2.9 logarithmic units (Table 3). H2TBA is the weakest base among all the ligands, while its analogues are stronger by approximately 0.7 logarithmic units. This characterizes them as very weak bases, which, even in strongly acidic solutions (C(H+) > 10 mol·L−1) have a marked fraction of the neutral form. The pK H values (and solvation coefficient m*) for H2DETBA and H2DBTBA are almost identical. For all three ligands m* ≈ 0.8, which is typical for ketones [51]. The similar values of m* give evidence of the similarity of the protonation mechanism for all three ligands in aqueous solution.
The complexation process of Ag+ with H2DETBA can be described by the equation:
The log10 K value obtained for the system is equal to 2.66 ± 0.01 (Table 4). This constant remains unchanged at each acidity. The obtained log10(ε) at 313 nm equals 3.77 ± 0.01. These data indicate that n in Eq. 7 is equal to 0. This also means that the AgH2DETBA monocomplex is formed through S-coordination. According to Eq. 7 with n = 0, stability constants can be obtained from this condition as [52]:
The obtained true stability constant for the monocomplex is small compared with other cyclic S-donor ligands. The log10 K values at 298 K and I = 0.5 are 1.95 mol·L−1 for thiozole [53], 4.26 for thiohydantion [53] and 8.84 for thiozolidine [53], respectively. This fact can be explained by the presence of steric hindrance through S-coordination due to the presence of ethyl groups in the 1,3 position. Maximum ΔA value for the Ag–H2DETBA system is at 310 nm. The values of the extinction are given in Table 3 at 313 nm for the convenience of calculation at this wavelength. According to [54], the Ag–H2DETBA complex species has S- and O-coordination in the solid state.
The experimental data for the Ag–H2DETBA system provides the possibility of formation of two monocomplex species coexisting in solution (Fig. 3). Quantum chemical calculations, dedicated to estimation of some thermodynamic parameters of protonation, dissociation and complex formation processes, were carried out based on cycles shown in Fig. 4 [55].

Optimize structure of AgH2DETBA: keto-isomer (a) and enol-isomer (b)

The thermodynamic cycles for calculation (from top to bottom): pK a, pK H and complex formation
These cycles were chosen as delivering the best results. Calculations of the acid–base properties in the pH-region were performed using the mono-solvated anion species (Fig. 5). For a clear estimation of equilibrium constants both specific and nonspecific solvation must be taken into account. In our case the contribution of specific solvation clearly prevails. Results obtained without taking into account specific solvation are at odds with experimental data by 3–5 orders of magnitude. Using in pK a calculations anions with any other coordinated water molecules (through nitrogen or sulphur atom) leads to results that have much larger discrepancies with experiment. Also, in anionic species, oxygens have the greatest negative charge, which makes this atom the most favorable for the formation of hydrogen bonds. Thus, accurate estimation of solvation Gibbs energies for these systems requires the explicit inclusion of solvent.

Optimized geometries of HTBA−·H2O (a), HDETBA−·H2O (b), and HDBTBA−·H2O (c)
The equilibrium constants were calculated using Eqs. 9–14 (for H3DETBA+ for example) [56]:
where
Here, RTln([H2O]) is a Gibbs energy change associated with moving a solvent from a standard state solution phase concentration of 1 mol·L−1 to a standard state of the pure liquid, 55.34 mol·L−1 [56, 57].
E zpe is the harmonic vibrational frequency calculated to estimate the zero point energy correction. Gibbs energies for ionization and solvation were used for calculations of ΔG both in the solid state and in solution. Values of G gas(H+) and ΔG solv.(H+) (−26.28 and −1108.27 kJ·mol−1 respectively) were taken from previous research [42]. Data for calculated values from Table 5 are presented in Supplementary Table S6. According to Table 5, a better approximation is observed for H2DBTBA. For others ligands, errors ranged from 0.2 to 0.6 logarithmic units. Besides, the strength of base increases in the order H2TBA < H2DBTBA < H2DETBA both for the experimental and calculated data. The values of pK a(calc.) obtained are in sharp disagreement with the experimental data (Table 3). For all ligands, the difference |pK a(calc.) − pK a(exp.)| is above 0.5 logarithmic unit. The maximum disagreement with the experiment was observed for H2TBA, the minimum for H2DBTBA. The calculated acidic strength decreases in the order H2DBTBA > H2DETBA > H2TBA, in complete disagreement with the experimental observations (Table 3). According to Table 5, calculations provide a better approximation for S-coordinated complex species (keto-isomer). Also, for S-coordinated complex species, the log10 K(calc.) value for keto-isomer is larger than that for enol-isomer more than on 1.2 logarithmic units and is close to the experimental value.
A TD–DFT study was carried out on the electronic structure of every species, to investigate charge transfer. Figure 6, for example, shows rendered HOMO and LUMO of H2DETBA for the neutral keto-form and HOMO/LUMO for Ag– H2DETBA. Other results for TD–DFT calculations are shown in Supplementary Figs. S14–S24 and Supplementary Tables S7 and S8.

Visualized HOMO (HOMO-3, HOMO-2, and HOMO-1) and LUMO for H2DETBA and Ag–H2DETBA complex species
For the anion, cation, enol and keto-forms there are two π–π* transitions from the sulfur atom of the thione group on the carbon atom (position 2) and the transition from the oxygen atom on the closest carbon atom (position 4) occur for all forms of all ligands: π(S) → π*(ring) + π(O) → π*(ring). For the S-coordinated complex, the TD–DFT (PBE0/SMD) results (Fig. 6; Supplementary Table S7) are in satisfactory agreement with the experimental data (λ exp 310 nm vs. 286 nm λ calc.). As in the case of the initial ligand, the Ag-complex reveals a π–π* transition: z 2(Ag) + π(S) → π*(ring).
Due to the weak interaction in [Ag(H2O)]H2DETBA, the results can be used for hydrothermal synthesis of the nanoparticles of Ag2S [58] and enhance anti-cancer activity of silver complexes.
4 Conclusion
The same mechanism for protonation of neutral molecules in highly acidic solutions is confirmed for all of the investigated ligands. According to ab initio simulations, protonation leads to formation of the enol tautomers through the oxygen atom protonation. The neutral form of ligand in solution is mostly in the keto-form. We show that the N,N-substituted thiobarbituric acid forms the S-coordinated monocomplex species with Ag(I). According to the TD–DFT calculations, π–π* transitions for all species were proposed. Ab initio calculations for thiobarbituric acid are acceptable to describe the properties of individual compounds, but accuracy of the description is decreased for general regularities.
References
da Marisco, C.M., Ribeiro, C.P., Bonini, J.S.: Ammonia potentiates methylmalonic acid-induced convulsions and TBARS production. Exp. Neurol. 182, 455–460 (2003)
Farias, I.L., Farias, J.G., Rossato, L., Araújo, M.C., Chiesa, J.: Correlation between TBARS levels and glycolytic enzymes: the importance to the initial evaluation of clinical outcome of colorectal cancer patients. Biomed. Pharmacother. 65, 395–400 (2011)
Hoda, A.A., Mostafa, I.E., Laila, A.R.: Thiobarbituric acid reactive substance (TBARS) a marker of oxidative stress in obstructive sleep apnea. Egypt. J. Chest Dis. Tuberc. 63, 119–124 (2014)
Barc, S., Stadelmann, S.I.: Effect of ischemia on TBARS and lactate production in several cerebral regions of anaesthetised and awake rats. Life Sci. 74, 3103–3113 (2004)
Ezzaher, A., Mouhamed, D.H., Mechri, A.: TBARs and non-enzymatic antioxidant parameters in Tunisian bipolar I patients. Immuno-Anal. Biol. Spéc. 27, 315–324 (2012)
Diaz, P., Linares, M.B., Egea, M., Linares, M.B.: Egea, M.: TBARs distillation method: Revision to minimize the interference from yellow pigments in meat products. Meat Sci. 98, 569–573 (2014)
Nowak, D., Katucka, S., Biatasiewicz, P.: Exhalation of H2O2 and thiobarbituric acid reactive substances (TBARs) by healthy subjects. Free Radic. Biol. Med. 30, 178–186 (2011)
Papastergiadis, A., Mubiru, E., Van Langenhove, H.: Malondialdehyde measurement in oxidized foods: evaluation of the spectrophotometric thiobarbituric acid reactive substances (TBARS) test in various foods. J. Agric. Food Chem. 60, 9589–9594 (2012)
de Azevedo, G., da Silva, E., Nascimento, M.R.: Evaluation of the 2-thiobarbituric acid method for the measurement of lipid oxidation in mechanically deboned gamma irradiated chicken meat. Food Chem. 80, 433–437 (2003)
Guillén-Sans, R.M., Guzmán-Chozas, N.: The thiobarbituric acid (TBA) reaction in foods: a review. Crit. Rev. Food Sci. Nutr. 38, 315–330 (1998)
Faidallah, H.M., Khan, K.A.: Synthesis and biological evaluation of new barbituric and thiobarbituric acid fluoro analogs of benzenesulfonamides as antidiabetic and antibacterial agents. J. Fluor. Chem. 142, 96–104 (2012)
Liang, M., Li, S., Zheng, H.: Synthesis and biological activity of novel barbituric and thiobarbituric acid derivatives against non-alcoholic fatty liver disease. Eur. J. Med. Chem. 46, 2003–2010 (2011)
Bloom, R.J., Westerfeld, W.W.: The thiobarbituric acid reaction in relation to fatty livers. Arch. Biochem. Biophys. 145, 669–675 (1971)
Suda, I., Furuta, S., Nishiba, Y.: Fluorometric determination of a 1,3-diethyl-2-thiobarbituric malondialdehyde adduct as an index of lipid peroxidation in plant. Biotechnol. Biochem. 58, 14–17 (1994)
Tabassum, S., Siddiqi, K.S., Khan, N.H., Kureshy, R.I.: Studies on complexes of thiobarbituric and 5,5-diethylbarbituric acid with lanthanide ions. Ind. J. Chem. 26, 489–495 (1987)
Moamen, S.R., El-Korashy, S.A., Ahmed, A.S.: A convenient method for the preparation of barbituric and thiobarbituric acid transition metal complexes. Spectrochim. Acta Part A 71, 1084–1094 (2008)
Masoud, M.S., El-Dessouky, M.A.: Transition metal (disubstitutedphenylazo)-barbituric and thiobarbituric acid complexes. Trans. Met. Chem. 15, 443–448 (1990)
Li, X., Wu, D., Gu, Y., Gan, F.: Synthesis, spectral and thermal properties of some transition metal(II) complexes with a novel ligand derived from thiobarbituric acid. Therm. Anal. Calorim. 98, 387–394 (2009)
Murphy, R.J., Svehla, G.: An analytical study of metal–thiobarbituric acid complexes. Anal. Chim. Acta 99, 115–124 (1978)
Balas, R., Verginadis, S., Geromichalos, G., Kourkoumelis, N., Male, L.: Synthesis, structural characterization and biological studies of the triphenyltin(IV) complex with 2-thiobarbituric acid. Eur. J. Med. Chem. 46, 2835–2844 (2011)
Zambrzycka, E., Godlewska-Zytkiewicz, B.: A new ion imprinted polymer based on Ru(III)–thiobarbituric acid complex for solid phase extraction of ruthenium(III) prior to its determination by ETAAS. Microchim. Acta 181, 1019–1027 (2005)
Golovnev, N.N., Molokeev, M.S., Vereshchagin, S.N.: The cis–trans isomer transformation, spectroscopic and thermal properties of Li, Na, K 1,3-diethyl-2-thiobarbiturate complexes. Polyhedron 85, 493–498 (2015)
Golovnev, N.N., Molokeev, M.S., Golovneva, I.: The crystal structure of lead(II) 1,3-diethyl-2-thiobarbiturate. Russ. J. Coord. Chem. 41, 300–304 (2015)
Loginovaa, N.V., Chernyavskayaa, G.I., Polozova, T.V.: Silver(I) interaction and complexation with sterically hindered sulfur-containing diphenol derivatives. Polyhedron 24, 611–618 (2005)
Lansdown, A.B.: A pharmacological and toxicological profile of silver as an antimicrobial agent in medical devices. Adv. Pharm. Sci. 2010, 1–16 (2010)
Eliseeva, S.V., Bunzli, J.-C.G.: Rare earths: jewels for functional materials of the future. New J. Chem. 35, 1165–1176 (2011)
Valiev, M., Bylaska, E.J., Govind, N., Kowalski, K., Straatsma, T.P.: NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 181, 1477–1489 (2010)
http://www.cluster.sfu-kras.ru (accessed April 1, 2015)
Adamo, C., Barone, V.: Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999)
Dunning Jr., T.H.: Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989)
Peterson, K.A., Puzzarini, C.: Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 114, 283–296 (2005)
Marenich, A.V., Cramer, C.J., Truhlar, D.G.: Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009)
Zhurko, G.A.: ChemCraft, version 1.7. http://www.chemcraftprog.com (accessed March 1, 2015)
Leggett, D.J.: Computational Methods for the Determination of Formation Constants. Plenum Press, New York (1985)
Tiwari, S., Ghosh, K.K.: Mixed micellization properties of cationic monomeric and gemini surfactants. J. Chem. Eng. Data 55, 4162–4167 (2010)
Cox, R.: Acidity functions: an update. Can. J. Chem. 61, 2225–2229 (1983)
Cox, R.: The excess acidity of aqueous HCl and HBr media. An improved method for the calculation of X-functions and H. Can. J. Chem. 59, 2023–2028 (1981)
Celik, H., Büyükaga, M.: Determination of pK a values of some benzoxazoline derivatives and the structure–activity relationship. J. Chem. Eng. Data 58, 1589–1596 (2013)
http://www.scilab.org/ (accessed July 1, 2015)
Grebenyuk, S.A., Perepichka, I.F., Popov, A.F.: Evaluation of the parameters of 1:1 charge transfer complexes from spectrophotometric data by non-linear numerical method. Spectrochim. Acta Part A 58, 2913–2923 (2002)
Mendez, E., Cerda, M.F., Gancheff, J.S.: Tautomeric forms of 2-thiobarbituric acid as studied in the solid, in polar solutions, and on gold nanoparticles. J. Phys. Chem. C 111, 3369–3383 (2007)
TellezSoto, C.A., Ramos, J.M., Junior, A.C.: Surface enhancement Raman scattering of tautomericthiobarbituric acid: natural bond orbitals and B3LYP/6311+G(d, p) assignments of the Fourier infrared and Fourier Raman spectra. Spectrochim. Acta A 114, 475–485 (2013)
Petrov, A.I., Lutoshkin, M.A., Taydakov, I.V.: Aqueous complexation of Y(III), La(III), Nd(III), Sm(III), Eu(III), and Yb(III) with some heterocyclic substituted β-diketones. Eur. J. Inorg. Chem. 6, 1074–1082 (2015)
Street, H.V., McMartin, C.: Action of concentrated sulphuric acid on 5,5-disubstituted barbituric acid derivatives. Clin. Chim. Actu. 9, 301–308 (1964)
Maynert, E.W., Washbur, E.: Reactions of barbituric acids in sulfuric acid. J. Am. Chem. Soc. 75, 700–704 (1953)
Singh, B.R., Jain, R.K., Jain, M.K., Ghosh, R.: Thermodynamics of the interaction of some transition metal ions with some N-substituted thiobarbituric acids. Thermochim. Acta 78, 175–180 (1984)
Smyth, W.F., Svehla, G., Zuman, P.: Polarographic and spectral investigation of acid–base equilibria in aqueous solutions of 2-thiobarbituric acids with substituents on sulphur. Anal. Chim. Acta 52, 129–138 (1970)
Izquerdo, A., Beltran, J., Guitras, L.: Thermodynamic properties of the dissociation of 2-mercaptopyridine, 2-mercaptopyrimidine and 2-thiobarbituric acid. Thermochim. Acta 127, 81–88 (1988)
Blais, M.S., Enea, O., Berthon, G.: Relations structure–réactivité grandeurs thermodynamiques de protonation geterocycles saturés et non saturés. Thermochim. Acta 20, 335–338 (1977)
Sansand, R.G., Chozas, M.G.: Spectrophotometric evaluation of ionization constants of some arylidene thiobarbituric acids. Microchem. J. 37, 40–50 (1988)
Cox, R.A., Clinton, R.S., Yates, K.: The excess acidity method. The basicities, and rates and mechanisms of enolization, of some acetoghenones and acetone, in moderately concentrated sulfuric acid. Can. J. Chem. 57, 2952–2959 (1979)
Burgot, J.-L.: Ionic Equilibria in Analytical Chemistry, pp. 452–458. Springer, Dordrecht (2012)
Blais, M.-J., Berthon, G.: Formation constants for complexes of silver with some heterocyclic ligands containing nitrogen and sulphur as donor atoms. J. Inorg. Nucl. Chem. 41, 933–935 (1979)
Golovnev, N.N., Molokeev, M.S., Lutoshkin, M.A.: Crystal structure of catena-(μ4-1,3-diethyl-2-thiobarbiturato-O, O′, S, S′) silver(I). Russ. J. Inorg. Chem. 60, 572–576 (2015)
Crugeiras, J., Ríos, A., Maskill, H.J.: DFT and AIM Study of the protonation of nitrous acid and the pK a of nitrous acidium ion. J. Phys. Chem. 115, 12357–12363 (2011)
Bryantsev, V.S., Mamadou, S.D., Goddard III, W.A.: Calculation of solvation free energies of charged solutes using mixed cluster/continuum models. J. Phys. Chem. B. 112, 9709–9719 (2008)
Vukovic, S., Hay, B.P., Bryantsev, V.S.: Predicting stability constants for uranyl complexes using density functional theory. Theor. Inorg. Chem. 54, 3995–4001 (2015)
Kumari, P., Chandran, P., Sudheer, K.S.: Synthesis and characterization of silver sulfide nanoparticles for photocatalytic and antimicrobial applications. J. Photochem. Photobiol. B 141, 235–240 (2014)
Acknowledgments
The research has been funded and carried out in terms of state contract (No. 3049) of Ministry of Education and Science of Russian Federation. The authors would also like to thank SFU CEJU for technical support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lutoshkin, M.A., Petrov, A.I. & Golovnev, N.N. Acid–Base, Complexing and Spectral Properties of Thiobarbituric Acid and Its 1,3-Derivatives in Aqueous Solutions: Spectrophotometric and Quantum Chemical Approach. J Solution Chem 45, 1453–1467 (2016). https://doi.org/10.1007/s10953-016-0525-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-016-0525-3