
Abstract
The stability and electronic and magnetic properties of RhnOs (n= 2–12) clusters in their most stable configurations were systematically studied by using density functional theory (DFT) at M06L/aug-cc-pVDZ level. Calculation of the second-order difference of energies and fragmentation energies exhibited that Rh3Os, Rh5Os, Rh7Os, and Rh9Os clusters are more stable than any other clusters. The calculated HOMO-LUMO energy gaps of the RhnOs clusters are found to be in the range of 0.018 to 0.299 eV, implying that the metallic behavior can appear in these clusters. Accordingly, the RhnOs clusters can be employed as heterogeneous nanocatalysts in many chemical reactions. The local Fukui function (\(f_{k}^{-} )\) has also been calculated, and the obtained results reveal that the highest \(f_{k}^{-} \) values are predicted for the Rh atoms. Therefore, the Rh atoms in the clusters are considered the most reactive sites that undergo reactions with electrophilic reagents. The analysis of the magnetic properties of the RhnOs clusters shows that the total magnetic moment per atom of these clusters varies from 0.67 to 1.75 µB/atom. And, the PDOS analysis reveals that the d orbitals play a crucial role for the magnetism of the RhnOs clusters, and the contribution of the s and p orbitals is small.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Over the last two decades, small transition metal clusters have received great attention due to their unique physical properties as compared with individual atoms, molecules, or bulk metal [1,2,3,4,5,6]. Hence, these clusters are widely applied in microelectronics, optic, magnetic, and nanotechnology [7,8,9,10,11]. They have also been extensively employed as nanocatalysts for various catalytic reactions [12,13,14,15]. For example, rhodium nanoclusters display excellent reactivity towards hydrogenation of aromatic compounds and have also been reported in the literature as the nanocatalysts of choice in hydroformylation of alkenes for the production of aldehydes [16,17,18,19,20,21,22]. The experimental observation exhibits that the catalytic performance of these clusters depends not only on the cluster size and shape but also on the cluster composition. Another interesting aspect is their remarkable magnetic properties, which shows that the small rhodium clusters are superparamagnetic at 93 K, with magnetic moments ranging from 0.35 to 1.09 µB/atom [23].
Osmium is also the most important element that plays an important role in many industrial processes; particularly, it is used as a catalyst for dehydrogenation of alcohols [24]. These heterogeneous osmium catalysts have shown high reactivity and good chemical stability in dehydrogenation of alcohols.
On the other hand, numerous experimental investigations were carried out to study the doped Rh clusters [25,26,27]. The reason is that addition of a second metal is a way to improve the structural, electronic, magnetic, and catalytic properties of the first one. For example, the molybdenum-doped rhodium clusters exhibited much higher catalytic activity than the pure one [26]. Similar results have been found for the tungsten-doped rhodium clusters. Recently, bimetallic RhPd clusters prepared by alcohol reduction method have also been employed as nanocatalysts for dehydrogenation of ammonia borane [28]. This molecule (ammonia borane) has been widely used today as new potential hydrogen storage materials due to its high hydrogen content (19.6 wt%) and high stability [29,30,31]. The same reaction (ammonia borane dehydrogenation) on the RhNi bimetallic clusters show excellent reactivity and durable stability [30]. In general, the doped rhodium clusters with the atoms of other elements exhibit markedly improved physical properties compared with that of pure rhodium clusters. On the theoretical side, we can say that there are very few theoretical investigations on the physical and chemical properties of TM-doped rhodium clusters during the last two decades [32,33,34,35]. Dennler et al. [34] investigated the structure and magnetic properties of RhnCom (n + m≤ 4) clusters and found that the presence of Co atoms in the cluster results in a remarkable increase of the local moments of Rh neighbors. Mokkath et al. [33] have studied the stabilities and magnetic and electronic properties of small FenRhm (n + m≤ 8) clusters employing the generalized gradient approximation (GGA). They found a significant enhancement of the local Fe moments as a result of Rh doping. Furthermore, the largest part of the spin polarization can mostly be traced back to the local d magnetic moments (about 90%), and the contribution of the s and p spin polarizations are almost negligible. Recently, manganese-doped rhodium clusters, RhxMny (x + y= 2–4), have been investigated using PBE/SDD method by Srivastava et al. [32]. They found that Rh2Mn2 cluster has the higher chemical stability than the other clusters. And, the magnetic properties of these clusters are strongly influenced by their proportion of Rh and/or Mn atoms. Using GGA-PW91 method, Lv et al. [35] investigated the equilibrium configurations and magnetic and electronic properties of CoxRh (x= 1–8) clusters and found that the Co2Rh, Co4Rh, and Co7Rh clusters are more stable than their neighbors. Further, the magnetism calculations show that the two atoms are aligned ferromagnetically in the CoxRh clusters and the total magnetic moment is mainly localized on the cobalt atom. Additionally, they also adopted identical method (GGA-PW91) to investigate the (RhCo)n (n≤ 5) clusters, and their results indicate that the cobalt-doped rhodium clusters can induce a significant change in the magnetic properties of the rhodium clusters. And, the local moment of Co atom has important improvement in (RhCo)n clusters (except for n= 2). This result is ascribed to the increase of the Rh–Co bond lengths in these binary clusters. Yang et al. [36] have studied the influence of Rh doping on the relative stabilities and electronic properties of Au nanoclusters. The results obtained by them show that the clusters with even number of atoms are more stable than those with odd number of atoms. Particularly, the Au5Rh cluster possesses a higher chemical stability than their neighboring nanoclusters, and thus can be considered a magic cluster.
In the present research, we optimize all the possible initial configurations of RhnOs (n= 2–12) clusters by employing density functional theory (DFT) approach. Based on the most stable geometries, the stability and electronic and magnetic properties of the RhnOs clusters have been calculated and analyzed. Further, the reactivity of each atom in these binary clusters has also been evaluated. The present paper is organized as: Section 2 which gives a brief description of the computational method employed in the present paper. Then detailed results and discussions of the stabilities and electronic and magnetic properties of the RhnOs clusters are described in Section 3. Finally, the conclusions are given in Section 4.
2 Theoretical Model
In this work, all calculations of the RhnOs (n= 2–12) clusters have been carried out using DFT provided by the Gaussian09 program [37]. We have chosen to use the meta-GGA functional M06L in our geometry optimization calculation for these clusters [38, 39]. Meanwhile, the aug-cc-pVDZ basis set is employed to describe the Rh and Os atoms [40]. This basis set became widely adopted in studying the equilibrium configurations, stabilities, and electronic properties of nanoclusters containing transition metal atoms [41,42,43].
In order to check the reliability of the method and basis set used in our calculations, we calculate the properties of Rh2 and Os2 dimers, respectively. The bond lengths (R), vibrational frequencies (ω), and binding energies (Eb) have been calculated and compared with available experimental data and other theoretical results (see Table 1).
Our optimizations predict a quintet spin state as the ground state of Rh2, which is well consistent with other theoretical works [44, 45]. The bond length (2.227 Å), vibrational frequency (317.2 cm− 1), and the binding energy (1.489 eV/atom) of Rh2 dimer were calculated, which are in better agreement with the experimental values of 2.28 Å, 267 cm− 1, and 1.46 eV/atom, respectively [46]. Further, our theoretical results are not only consistent with the experimental values but are also in excellent agreement with previous studies [44, 45].
The Os2 dimer has a ground state (7Σ) with a bond length of 2.256 Å and vibrational frequency of 274.4 cm− 1, which are in good agreement with the experiment data (see Table 1) [47, 48]. The calculated binding energy of Os2 dimer (1.51 eV/atom) was found to be smaller than the experimentally derived value (2.15 eV/atom). Moreover, our results fit well with the other theoretical calculations [49, 50]. Du et al. [50] obtained a bond length and binding energy per atom of 2.284 Å and 1.26 eV/atom, respectively, using the B3LYP/LANL2DZ method. Wu et al. [49] predicted a bond length of 2.238 Å, and binding energy per atom of 1.25 eV/atom, using B3LYP method and CEP-121G basis set.
In order to obtain the equilibrium configurations of the RhnOs (n= 2–12) clusters, a lot of possible initial geometries, which include one-, two-, and three-dimensional geometries with different spin states have been tested. The number of possible initial geometries increases rapidly with the increase in size of clusters. For example, the number of initial candidate configurations of the Rh7Os cluster is 34, whereas for the Rh3Os cluster, the number of possible initial geometries is only 12. Moreover, in our calculations, all the optimized configurations with the energy minimum have been verified by computing the harmonic vibrational frequencies without imaginary mode.
3 Results and Discussion
3.1 Equilibrium Configurations of RhnOs Clusters
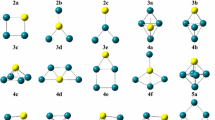
In this study, we have optimized a large number of initial configurations for each cluster size. The results obtained for the most stable geometries and some low-lying energy isomers of the RhnOs (n= 2–12) clusters are illustrated in Figs. 1 and 2. Their corresponding relative energies (ΔE), binding energies (Eb), spin multiplicities (M), point-group symmetries (PG), average bond lengths (R), and total magnetic moments per atom (µT) are shown in Table 2. All optimized isomers were labeled as na, nb, nc, nd, ne, and nf, where n represents the number of Rh atoms in the binary RhnOs clusters, and the configurations labeled in ‘na’ correspond to the most stable geometries.

The lowest energy and low-lying structures of RhnOs (n= 2–7) clusters. n a, lowest-energy geometries; n b–n f, few low-lying geometries. The green and brown balls represent the Rh and Os atoms, respectively

The lowest-energy and low-lying structures of RhnOs (n= 8–12) clusters
As shown in Figs. 1 and 2, the lowest-energy structures of the RhnOs clusters prefer the three-dimensional configurations, except for very small cluster sizes (n≤ 3). Further, the Os atom prefers to adsorb on the surface sites in the cluster (except for Rh12Os cluster). The obtained results for RhOs cluster indicate that the sextet spin state is lower in energy than the quartet and octet spin states by 0.68 and 1.18 eV, respectively. Accordingly, the sextet RhOs cluster with electronic state 6Σ is the most stable configuration. Also, the results show that the Rh–Os bond length is 2.242 Å, which is slightly larger than the Rh–Rh bond (2.227 Å) in Rh2 cluster. The corresponding binding energy per atom of this cluster is 1.735 eV/atom. To our knowledge, there have been no experimental data on RhOs dimer to which these results could be directly compared.
For the Rh2Os, the ground state geometry is an isosceles triangle (2a) with C2V symmetry and quintet spin multiplicity (5B2), which has two Rh–Os bonds of 2.315 Å and one Rh–Rh bond of 2.734 Å, respectively. The 2b isomer is a linear geometry with Os atom at the middle. The corresponding electronic state is 3Ξg. Its energy is less stable than the lowest-energy configuration (2a) by 0.87 eV.
The 2c isomer in quintet spin state is found to form planar geometry. This geometry is energetically higher than the most stable configuration by 1.99 eV.
The lowest-energy structure of the Rh3Os cluster is an irregular triangle pyramid geometry (3a) with electronic state of 8A″ and CS symmetry. Similar configuration was also obtained as the next stable isomer (3b). Its energy is only 5 ×10− 4 eV less stable than the ground-state geometry. Therefore, the configurations 3a and 3b should be considered to be degenerate. The rhombus configuration (isomer 3d) with an electronic state of 4B1 was predicted as a low-lying geometry with only 1.14 eV higher in energy compared with the most stable geometry (3a). Two Y-shaped isomers (3c and 3e) with the same symmetry (C2V) have also been optimized, and the energy difference between these two geometries (3c and 3e) is 1.33 eV.
The most stable structure of the Rh4Os cluster is a triangular bipyramid geometry with an Os atom at the middle plane (see isomer 4a). The symmetry and electronic state of this cluster are C2V and 7A1, respectively. Identical configuration (except that the Os atom is localized on the vertex) has also been optimized in our study (4c). This configuration is higher in energy than the ground-state geometry by only 0.07 eV. The second low-lying isomer is a square pyramid with an Os atom at the bottom (4b). Its total energy is only 0.05 eV lower than the isomer 4c. The next isomer 4d is a planar trapezoidal geometry with the Os centered in the middle of bottom lateral. The isomer 4e is also a planar geometry, and its energy is 0.79 eV higher than the isomer 4d.
In this case, the lowest-energy geometry of the Rh5Os cluster is a distorted octahedron with the Os located at the apex of the octahedron (5a). The corresponding symmetry is C4V. The second lowest-energy configuration 5b is a prism. It is found to be 0.67 eV higher in energy than the lowest-energy geometry (5a). The isomer 5c is the third low-lying structure, which is a capped trigonal bipyramid geometry (5c), being 0.37 eV higher in energy than the isomer 5b. The next isomer 5d is a capped square pyramid configuration with the Os atom being localized at the vertex of the pyramid. A similar configuration with an Os atom capping one of the triangular faces (5e) was found as a low-lying structure. The difference of energy between 5d and 5e is only 0.22 eV. The planar geometry (isomer 5f) has also been optimized in our calculations, and its energy is about 1.01 eV higher in energy than the isomer 5e.
For the Rh6Os cluster, the distorted octahedron geometry of which a face is capped by Rh atom was predicted as the ground-state geometry (6a). The symmetry and electronic state of this stable cluster are CS and 11A″, respectively. Isomer 6b, which is almost identical to the configuration 6a (capped octahedron geometry), is higher in energy than the ground-state geometry by only 0.36 eV. Another three-dimensional (3D) configuration, isomer 6c was also identified as low-lying structure in our optimizations. This configuration is about 0.60 eV higher in energy than the most stable geometry (6a). The next stable isomer 6d is a capped triangular prism structure. Its energy is 0.70 eV higher than the most stable configuration (6a). The next stable isomer 6e is a planar geometry, and its energy is 0.94 eV higher than the ground-state geometry (6a).
The most stable geometry of the Rh7Os cluster is a bicapped octahedron structure (C2V symmetry) with Os atom in the vertex site of the octahedron (7a). Its corresponding electronic state is 10A″. The next isomer 7b is also a bicapped octahedron with an Os atom in the middle plane of the octahedron. The next configuration can be viewed as a fusion of two prisms (7c), and its total energy is higher than the most stable configuration (7a) by 1.23 eV. In our calculations, the identical geometry (7d) to that of the most stable structure has also been identified as the metastable geometry, in which the Os atom lies at the one vertex site of the octahedron, while two capped Rh atoms are located at both sides of the other vertex atom of the octahedron. The difference in energy between this isomer and the most stable geometry is 1.57 eV. The next stable geometry 7e can be seen as a combination of two square pyramids (see Fig. 2). Its energy is less stable than 7d isomer by 1.03 eV. A similar configuration (7f) has also been predicted in our study, but being higher in energy than the lowest-energy geometry by 2.73 eV.
In the case of the Rh8Os cluster, all the optimized structures adopt 3D configurations. The most stable geometry can be viewed as a fusion of two octahedron configurations with C2V symmetry. The corresponding electronic state is 7A1. The next stable isomer found has a capped cubic configuration (8b). Identical configuration (8d) to the isomer 8b has also been predicted in our optimizations, and its energy is less stable than 8b isomer by 0.61 eV. The isomer 8c is obtained by adding two Rh atoms on the sides of 6d isomer. A 3D geometry has also been identified (8e) as the low-energy configuration, and its energy is only 0.09 eV higher than the isomer 8d.
For the Rh9Os cluster, the most stable structure can be described as a combination of two octahedron geometries (slightly distorted) with C2V symmetry (9a). The electronic state predicted for this stable configuration is 16A1. The isomer 9b was considered as a fusion of two trigonal prisms. Its total energy is only 0.70 eV higher than that of the lowest-energy structure. Isomer 9c, which is identical to the configuration 9b, is higher in energy than that of the most stable configuration by 0.71 eV. Hence, the energy difference between the two isomers (9b and 9c) is only 0.01 eV, indicating that the two optimized configurations are nearly degenerate. We also obtained a tetracapped octahedron configuration (9d) as a metastable isomer in our optimizations. It is found to be 0.47 eV higher in energy than the isomer 9c. In addition to that, a bicapped cubic structure (9e) was also predicted, and its energy is 3.10 eV higher than the most stable configuration (9a).
The ground-state structure of Rh10Os is viewed as a tricapped cubic geometry (10a) with C1 symmetry, and the value of spin multiplicity is 15. The second stable isomer (10b) is considered a fusion of one cubic and a capped trigonal prism, and its energy is less stable than the lowest-energy geometry by 1.51 eV. The third isomer (10c) is also a three-dimensional geometry. This configuration can be described as a fusion of a trigonal prism and a capped cubic with CS symmetry. The isomer 10d has been considered as a fusion of one cubic and a bicapped square pyramid. The last isomer (10e), in the size range, is a fusion of three square pyramids. This geometry was found to be higher in total energy by 0.70 and 0.81 eV than the geometries 10d and 10c, respectively.
For Rh11Os cluster, we have considered four kinds of geometry (11a–d) that represent the most stable configurations among the geometric configurations of all clusters optimized in our study. In this case, the most stable configuration is viewed as two cubic geometries fused on a square face with C2V symmetry and characterized by a high multiplicity (M= 12). Identical structure has also been predicted as the third low-lying geometry (11c), and its energy is higher than the ground-state structure by 0.47 eV. The second structure (11b) is considered a combination of one cubic and two trigonal prisms (11b). This isomer is energetically lower than the isomer 11c by 0.36 eV. The structure obtained by geometry optimization of isomer 11d is identical to that of the isomer 11b, and their energy is higher than the energy of isomer 11b by 1.12 eV.
In the case of Rh12Os cluster, the configuration is more stable when an Os atom is at the center of the cluster (see Fig. 2). The distorted icosahedron with the center doping Os atom is obtained as the most stable configuration (12a). The symmetry and electronic state of this isomer are C1 and 19A, respectively. The next stable isomer 12b can also be considered as an icosahedron with the Os atom at the apex position of the pentagon pyramid (12b). Similar configuration with the Os atom on the surface (12c) has also been predicted in this study. This configuration has been found to be less stable than the isomer 12b by only 0.01 eV. Hence, the two configurations (12b and 12c) are nearly degenerate in energy. The fourth geometry (isomer 12d) is also a three dimensional geometry, and its total energy is largely higher than the geometry 12a by 2.67 eV.
3.2 Stabilities of RhnOs Clusters
In order to understand the stabilities of the lowest-energy configurations of the RhnOs clusters, the fragmentation energies (ΔEf) and the second-order differences of the total energies (Δ2E) and the binding energies per atom (Eb) of different cluster sizes were calculated as follows:
where E(RhnOs), E(Rhn+ 1Os), and E(Rhn− 1Os) represent the total energies of the ground-state structure of the RhnOs, Rhn+ 1Os, and Rhn− 1Os clusters, respectively. E(Rh) and E(Os) represent the total energies of the Rh and Os atoms, respectively.
In nanocluster physics, the fragmentation energy and the second difference in energy are key parameters for examining the relative stability of the nanoclusters. Further, the values of Δ2E can be correlated well with the relative abundances estimated in mass spectroscopy experiments.
The binding energies (Eb) of the lowest-energy RhnOs clusters as a function of cluster size are-shown in Fig. 3. The second-order difference of energies (Δ2E) of these clusters has also been displayed in Fig. 4. Figure 3 shows that the binding energy per atom increases with the increasing of cluster size from n= 2 to n= 12, indicating that the RhnOs clusters can continuously gain energy during the growth process. From Fig. 4, the local peaks are found at n= 3, 5, 7, and 9, which indicates that the Rh3Os, Rh5Os, Rh7Os, and Rh9Os clusters are relatively more stable than any other nanoclusters. Therefore, these stable clusters are expected to have relatively high abundances in mass spectrometric measurements.

Binding energy per atom of the lowest-energy structures of RhnOs clusters as a function of cluster size

Size dependence of the second-order energy difference (Δ2E) of RhnOs clusters
We have also investigated the stability of these clusters by examining the fragmentation energy (ΔEf) as a function of the cluster size (Fig. 5). As can be seen from the results in Fig. 5, the local maxima occur at the Rh5Os, Rh7Os, and Rh9Os, which reflects that these clusters are remarkably stable than their corresponding neighbors. This result is in excellent agreement with the above analysis on the second-order difference of energies.

Size dependence of the fragmentation energies (ΔEf) of RhnOs clusters
3.3 Reactivity and Electronic Properties of RhnOs Clusters
The HOMO-LUMO gap is an important parameter to reflect the chemical reactivity of nanoclusters and their reactions with small molecules. A small HOMO-LUMO gap indicates a higher chemical reactivity of these clusters. The calculated HOMO-LUMO gaps of the RhnOs clusters are found to be in the range of 0.018 to 0.299 eV (see Table 3). Smaller HOMO-LUMO gap was observed for the Rh8Os cluster, indicating a great chemical reactivity. Further, the small HOMO-LUMO energy gaps indicate a metallic character of the RhnOs clusters. Hence, these clusters hold promise for design of new catalytic nanomaterials. Accordingly, the RhnOs clusters can be used as catalysts for many catalytic reactions, especially for selective hydrogenation reactions [16, 51]. They can also be employed as catalysts for dehydrogenation of ammonia borane and methylamine borane [29, 30].
The dipole moments (α) of the RhnOs clusters have been calculated, and the results are reported in Table 3. As can be seen from this table, the RhnOs clusters are characterized by small dipole moments, which vary between 0.039 and 1.01 debye. This is mostly due to the fact that the electronegativity of rhodium is about 2.28, which is almost equal to that of osmium (2.2). VIP and VEA for the lowest-energy RhnOs clusters have also been computed (see Table 3). The values of VEA were found to vary between 0.895 and 2.511 eV, and the calculated values of VIP range between 5.576 and 6.408 eV. Unfortunately, there are no experimental data for these clusters to compare our theoretical results. In addition, the VEA values are much lower than the VIP values, suggesting that these clusters can reflect the ability to gain electrons.
In this study, the chemical reactivity of each atom in the binary RhnOs clusters has also been evaluated using the condensed Fukui function, \(f_{k}^{-} \). This function \(f_{k}^{-} \) for electrophilic attack is defined as:
where qk (N) and qk (N − 1) present the charge of k atom in a cluster with N and N − 1 electrons, respectively.
The Fukui function is able to predict the possible binding site of a molecule on a metal cluster. Therefore, this function reflects the ability for clusters to undergo activated chemical reactions with small molecules.
The condensed Fukui function \(f_{k}^{-} \) for the different sites of the RhnOs clusters is listed in Table 4. For each cluster studied, the atoms of higher values of \(f_{k}^{-} \) are considered active sites where the chemical reactions occur. The atoms of lower values of \(f_{k}^{-} \) are considered inactive sites. Thus, the most reactive sites of these clusters are those with a large positive value of \(f_{k}^{-} \). From Table 4, it is clearly seen that the highest \(f_{k}^{-} \) values are predicted for the Rh atoms (except for the Rh5Os and Rh12Os clusters), and the Os atom in the same cluster was observed to be less active than the Rh atoms. This result indicates that the Rh atoms in the RhnOs clusters are more favorable to react with an electrophilic reagent. Hence, the rhodium atoms can be considered the most reactive sites that undergo chemical reactions with electrophilic reagents. In other words, the active site rhodium atoms in the RhnOs clusters can play a significant role in the elucidation of reaction mechanisms.
3.4 Magnetic Properties of RhnOs Clusters
The magnetic moments per atom of the lowest-energy RhnOs clusters as function of its size are illustrated in Fig. 6. The magnetic moment per atom exhibits irregular oscillating behavior, and the µT calculated for the RhnOs clusters are in the range 0.67–1.75 µB/atom. The Rh5,9Os and Rh3Os clusters present high magnetic moment corresponding to 1.50 and 1.75 µB/atom, respectively. The results also exhibit that the magnetic moment of these clusters strongly depends on their geometries. For example, the octahedron (5a) and capped square pyramid (5d) configurations have a magnetic moment of 1.5 and 0.5 µB/atom, respectively.

Size dependence of the total magnetic moment for the lowest-energy structures of RhnOs clusters
In order to further study the magnetic properties of the most stable configurations of the RhnOs clusters, we have performed the partial density of states (PDOS) from the contribution of different state components (s, p, and d orbitals). The results of PDOS for the RhnOs (n= 3, 5, 6, 7, 8, and 11) are shown in Fig. 7. The spin-up states is plotted as positive value, and spin-down states as negative. The Fermi level (EF) of the RhnOs clusters is presented as dashed vertical line (The calculated values for each cluster were reported in Table 3). As can be seen from Fig. 7, the magnetic properties below the Fermi level mostly originate from d states, and the contributions from s and p are very small. For example, in the Rh6Os cluster which possesses high magnetic moment (µT= 1.42 µB/atom), the contribution of d states to the magnetic moment is about 95%, while that from the s and p orbitals is only 5%.

The PDOS for the RhnOs (n= 3, 5, 6, 7, 8, and 11) clusters in their ground states
4 Conclusions
The stability and electronic and magnetic properties of the RhnOs clusters have been investigated at M06L level of theory using aug-cc-pVDZ basis set. The main findings of this work are summarized as follows:
-
(1)
The optimization calculations show that the 3D configurations of the RhnOs clusters are energetically much stable than the other two dimensions (2D). Furthermore, it was found that the Os atom prefers to adsorb on the surface of the cluster (except for Rh12Os cluster).
-
(2)
The second-order difference energy and the fragmentation energy of the RhnOs clusters exhibit that the Rh3Os, Rh5Os, Rh7Os, and Rh9Os clusters are energetically more stable than their neighboring clusters. The calculated HOMO-LUMO energy gaps of these clusters are in the range 0.018–0.299 eV. Smaller HOMO-LUMO gap is observed for the Rh8Os, indicating a much greater chemical reactivity of this cluster compared with other clusters. The results indicate also a metallic behavior for the RhnOs clusters, and thus these nanoclusters can be used as nanocatalysts for many catalytic reactions.
-
(3)
Fukui function (\(f_{k}^{-} )\) for electrophilic attack for the most stable geometries of the RhnOs clusters has been examined, and the results clearly predict that the Rh atoms in the cluster have higher values \(f_{k}^{-} \) for electrophilic attack. Meanwhile, the Os atom in the cluster was found to be less active than the rhodium atoms. Accordingly, the RhnOs clusters are more favorable to react with an electrophilic reagent.
-
(4)
The calculated magnetic properties exhibit that the magnetic moment of the RhnOs clusters was found to be in the range 0.67–1.75 µB/atom. Higher magnetic moment was observed for the Rh3Os cluster. The magnetic moment of these clusters was also found to be strongly influenced by the geometry of the cluster. For example, the calculated magnetic moment of the octahedron geometry (1.50 µB/atom) is about five times higher than that of the capped square pyramid geometry (0.50 µB/atom).
-
(5)
The PDOS analysis reveals that the d orbitals play a crucial role for the magnetism of the RhnOs clusters, and the contribution of the s and p orbitals to the magnetic moment is little.
References
Schmid, G.: Large clusters and colloids. Metals in the embryonic state. Chem. Rev. 92, 1709–1727 (1992)
Lewis, L.N.: Chemical catalysis by colloids and clusters. Chem. Rev. 93, 2693–2730 (1993)
Xu, X.S., Yin, S.Y., Moro, R., de Heer, W.A.: Magnetic moments and adiabatic magnetization of free cobalt clusters. Phys. Rev. Lett. 95, 237209 (2005)
Parks, E.K., Klots, T.D., Riley, S.J.: Chemical probes of metal cluster ionization potentials. J. Chem. Phys. 92, 3813–3826 (1990)
Cox, A.J., Louderback, J.G., Apsel, S.E., Bloomfield, L.A.: Magnetism in 4d-transition metal clusters. Phys. Rev. B 49, 12295–12298 (1994)
Soltani, A., Boudjahem, A.: Stabilities, electronic and magnetic properties of small Rhn (n = 2-12) clusters: a DFT approach. Comput. Theor. Chem. 1047, 6–14 (2014)
Yonezawa, T., Imamura, K., Kimizuka, N.: Direct preparation and size control of palladium nanoparticle hydrosols by water-soluble isocyanide ligands. Langmuir 17, 4701–4703 (2001)
Zhang, J.Y., Fang, Q., Kenyon, A.J., Boyd, I.W.: Visible photoluminescence from nanocrystalline Ge grwn at room temperature by photo-oxidation of SiGe using a 126 nm lamp. Appl. Surf. Sci. 208–209, 364–368 (2003)
Dong, C.D., Gong, X.G.: Magnetism enhanced layer-like structure of small cobalt clusters. Phys. Rev. B 78, 020409–020412 (2008)
Teranishi, T., Miyake, M.: Size control of palladium nanoparticles and their crystal structures. Chem. Mater. 10, 594–600 (1998)
Gopidas, K.R., Whitesell, J.M., Fox, M.A.: Synthesis, characterization, and catalytic applications of a palladium-nanoparticles-cored dendrimer. Nano. Lett. 3, 1757–1760 (2003)
Boudjahem, A., Chettibi, M., Monteverdi, S., Bettahar, M.: Acetylene hydrogenation over NiCu nanoparticles supported on silica prepared by aqueous hydrazine reduction. J. Nanosci. Nanotechnol. 9, 3546–3554 (2009)
Boudjahem, A., Redjel, A., Mokrane, T.: Preparation, characterization and performance of Pd/SiO2 catalyst for benzene catalytic hydrogenation. J. Ind. Eng. Chem. 18, 303–308 (2012)
Chettibi, M., Boudjahem, A., Bettahar, M.: Synthesis of Ni/SiO2 nanoparticles for catalytic benzene hydrogenation. Transit. Metal. Chem. 36, 163–169 (2011)
Boudjahem, A., Bouderbala, W., Bettahar, M.: Benzene hydrogenation over Ni-Cu/SiO2 catalysts prepared by aqueous hydrazine reduction. Fuel. Process. Technol. 92, 500–506 (2011)
Sidhpuria, K.B., Patel, H.A., Parikh, P.A., Bahadur, P., Bajaj, H.C., Jasra, R.V.: Rhodium nanoparticles intercalated into montmorillonite for hydrogenation of aromatic compounds in the presence of thiophene. Appl. Clay. Sci. 42, 386–390 (2009)
Sanchez, A., Fang, M., Ahmed, A., Sanchez-Delgado, R.A.: Hydrogenation of arenes, N-heteroaromatic compounds, and alkenes catalyzed by rhodium nanoparticles supported on magnesium oxide. Appl. Catal. A 477, 117–124 (2014)
Campos, C.H., Rosenberg, E., Fierro, J.L., Urbano, B.F., Rivas, B.L., Torres, C.C., Reyes, P.A.: Hydrogenation of nitro-compounds over rhodium catalysts supported on poly(acrylic acid)/Al2O3 composites. Appl. Catal. A 489, 280–291 (2015)
Behr, A., Brunsch, Y., Lux, A.: Rhodium nanoparticles as catalysts in the hydroformylation of 1-dodecene and their recycling in thermomorphic solvent systems. Tetrahedron. Lett. 53, 2680–2683 (2012)
Bruss, A.J., Gelesky, M.A., Machado, G., Dupont, J.: Rh(0) nanoparticles as catalyst precursors for the solventless hydroformylation of olefins. J. Mol. Catal. A 252, 212–218 (2006)
Yoon, T.J., Kim, J.I., Lee, J.K.: Rh-based olefin hydroformylation catalysts and the change of their catalytic activity depending on the size of immobilizing supporters. Inorg. Chim. Acta. 345, 228–234 (2003)
Han, D., Li, X., Zhang, H., Liu, Z., Hu, G., Li, C.: Asymmetric hydroformylation of olefins catalyzed by rhodium nanoparticles chirally stabilized with (R)-BINAP ligan. J. Mol. Catal. A 283, 15–22 (2008)
Cox, A.J., Louderback, J.G., Bloomfield, L.A.: Experimental observation of magnetism in rhodium clusters. Phys. Rev. Lett. 71, 923–926 (1993)
Bertoli, M., Choualeb, A., Lough, A.J., Moore, B., Spasyuk, D., Gusev, D.G.: Osmium and ruthenium catalysts for dehydrogenation of alcohols. Organometallics 30, 3479–3482 (2011)
Mendes, F.M., Schmal, M.: The cyclohexanol dehydrogenation on Rh-Cu/Al2O3 catalysts: chemisorpion and reaction. Appl. Catal. A: Gen. 163, 153–164 (1997)
Trunschke, A., Ewald, H., Gutschick, D., Miessner, H., Skupin, M., Walther, B., Bottcher, H.C.: New bimetallic Rh-Mo and Rh-W clusters as precursors for selective heterogeneous CO hydrogenation. J. Mol. Catal. 56, 95–106 (1989)
Zitoun, D., Amiens, C., Chaudret, B.: Synthesis and magnetism of CoxRh1−x and CoxRu1−x nanoparticles. J. Phys. Chem. B 107, 6997–7005 (2003)
Rakap, M.: The highest catalytic activity in the hydrolysis of ammonia borane by poly(N-vinyl-2-pyrrolidone)-protected palladium-rhodium nanoparticles for hydrogen generation. Appl. Catal. B 163, 129–134 (2015)
Jiang, H.-L., Xu, Q.: Catalytic hydrolysis of ammonia borane for chemical hydrogen storage. Catal. Today 170, 56–63 (2011)
Shen, J., Cao, N., Liu, Y., He, M., Hu, K., Luo, W., Cheng, G.: Hydrolytic dehydrogenation of amine-boranes catalyzed graphene supported rhodium-nickel nanoparticles. Catal. Commun. 59, 14–20 (2015)
Durap, F., Zahmakiran, M., Ozkar, S.: Water soluble laurate-stabilized rhodium (0) nanoclusters catalyst with unprecedented catalytic lifetime in the hydrolytic dehydrogenation of ammonia borane. Appl. Catal. A 369, 53–59 (2009)
Srivastava, A.K., Misra, N.: Structures, stabilities, electronic and magnetic properties of small RhxMny (x + y = 2-4) clusters. Comput. Theor. Chem. 1047, 1–5 (2014)
Mokkath, J.H., Pastor, G.M.: First-principles study of structural, magnetic, and electronic properties of small Fe-Rh alloy clusters. Phys. Rev. B 85, 054407 (2012)
Dennler, S., Morillo, J., Pastor, G.M.: Calculation of magnetic and structural properties of small Co-Rh clusters. Surf. Sci. 532–535, 334–340 (2003)
Lv, J., Bai, X., Jia, J.F., Xu, X.H., Wu, H.S.: Structural, electronic and magnetic properties of ConRh clusters from density functional calculations. Physica B. 407, 14–21 (2012)
Yang, J.X., Wei, C.F., Guo, J.J.: Density functional study of AunRh (n = 1-8) clusters. Physica B. 405, 4892–4896 (2010)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A. Jr., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Keith, T., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian 09, revision D.01. Gaussian, Inc., Wallingford (2013)
Zhao, Y., Truhlar, D.G.: A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 125, 194101 (2006)
Zhao, Y., Truhlar, D.G.: Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41, 157–167 (2008)
Dunning, T.H.: Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989)
Khetrapal, N.S., Jian, T., Lopez, G.V., Pande, S., Wang, L.-S., Zeng, X.C.: Probing the structural evolution of gold-aluminum bimetallic clusters (Au2Aln−, n = 3-11) using photoelectron spectroscopy and theoretical calculations. J. Phys. Chem. C 121, 18234–18243 (2017)
Khetrapal, N.S., Jian, T., Pal, R., Lopez, G.V., Pande, S., Wang, L.-S., Zeng, X.C.: Probing the structures of gold-aluminum alloy clusters AuxAly−: a joint experimental and theoretical study. Nanoscale 8, 9805–9814 (2016)
Khetrapal, N.S., Satya, S.S., Zeng, X.C.: Structural evolution of gold clusters Aun− (n = 21-25), revised. J. Phys. Chem. A 121, 2466–2474 (2017)
Beltran, M.R., Zamudio, F.B., Chauhan, V., Sen, P., Wang, H., Ko, Y.J., Bowen, K.: Ab initio and anion photoelectron studies of Rhn (n = 1-9) clusters. Eur. Phys. J. D 67, 63–70 (2013)
Chien, C.H., Blaisten-Barojas, E., Pederson, M.R.: Magnetic and electronic properties of rhodium clusters. Phys. Rev A 58, 2196–2202 (1998)
Gingerich, K.A., Cocke, D.L.: Thermodynamic confirmation for the high stability of gaseous TiRh as predicted by the Brewer-Engel metallic theory and the dissociation energy of diatomic rhodium. J. Chem. Soc. Chem. Commun. 1, 536–536 (1972)
Jules, J.L., Lombardi, J.R.: Transition metal dimer internuclear distances from measured force constants. J. Phys. Chem A 107, 1268–1273 (2003)
Morse, M.D.: Clusters of transition-metal atoms. Chem. Rev. 86, 1049–1109 (1986)
Du, J., Sun, X., Wang, H.: The confirmation of accurate combination of functional and basis set for transition-metal dimers: Fe2, Co2, Ni2, Ru2, Rh2, Pd2, Os2, Ir2, and Pt2. Int. J. Quantum. Chem. 108, 1517–1517 (2008)
Wu, Z.J., Han, B., Dai, Z.W., Jin, P.C.: Electronic properties of rhenium, osmium and iridium dimmers by density functional methods. Chem. Phys. Lett. 403, 367–371 (2005)
Cimpeanu, V., Kocevar, M., Parvulescu, V., Leitner, W.: Preparation of rhodium nanoparticles in carbon dioxide induced ionic liquids and their application to selective hydrogenation. Angew. Chem. Int. 48, 1085–1088 (2009)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Boudjahem, AG., Boulbazine, M. & Chettibi, M. Electronic and Magnetic Properties of Os-Doped Rhodium Clusters: a Theoretical Study. J Supercond Nov Magn 31, 3119–3131 (2018). https://doi.org/10.1007/s10948-018-4579-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-018-4579-x