
Abstract
Using the density functional theory (DFT), the geometries, relative stabilities and magnetic properties of bimetallic RhnOs (n = 1–9) clusters have been investigated. The relative stability was analyzed by examining the binding energy, fragmentation energy, second-order differences of energies and HOMO–LUMO energy gaps. The obtained results indicate that RhOs, Rh3Os, Rh5Os and Rh7Os clusters are more stable than their neighboring clusters. In addition, the doping of the Os atom enhanced the stability of the Rh clusters. The chemical hardness and chemical potential show that RhOs cluster is less reactive, indicating that RhOs cluster is the most stable one among all the clusters. The magnetic properties calculations exhibited that total magnetic moments come mostly from the Rh atoms for RhnOs (n = 3–9) clusters, while the contribution of the Os atom is observed for RhOs and Rh2Os clusters. In addition, the d orbitals plays an important role in the magnetic moments of the RhnOs clusters.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
During these last years, transition metal clusters received a big attention because of their specific properties in comparison with those of individual atoms or bulk metals [1–6]. The rhodium clusters are used extensively in many applications as electronic, optic, magnetic and the nanotechnology [7–11]. They have also been the choice as catalysts in many catalytic reactions, notably for the hydrogenation of the aromatic compounds and the hydroformylation of alkenes for the production of aldehydes [12–19]. In addition, the experimental observations show that the catalytic activity of these clusters depends on the size, on the structure and the morphology of the clusters [13–15]. Rhodium clusters have a very interesting magnetic properties, and the experience shows that these clusters are superparamagnetic at very low temperature (93 K) [20]. On the other hand, the osmium clusters have been used in several catalytic applications, and many experimental results indicate that osmium clusters present an important chemical reactivity [21–23]. For example, osmium catalysts has been found to be very efficient for the reaction of methane dissociation [22]. Another work shows that the addition of osmium in carbon nanotube increase the stability and improve the reactivity of the carbon nanotube [23, 24].
The reason of adding a second metal to the rhodium clusters is to improve the physical and chemical properties of these last. Indeed, several experimental studies have been investigated the effect of the doping on the structural, electronic and catalytic properties of rhodium clusters [25–27]. The experimental observations show that the rhodium clusters doped with the molybdenum and tungsten present an important catalytic activity than those of the monometallic clusters [26]. In general, the doping of the rhodium clusters with transition metals atoms improve the physical and chemical properties of the pure rhodium clusters.
On the other hand, our bibliographic research indicate that there are few theoretical works concerning the structural, electronic and magnetic properties of the rhodium clusters doped by transition metal atoms [28–33]. For example, Dennler and al. [28] studied the structural and magnetic properties of the binary RhxCoy (x + y ≤ 4) clusters. The obtained results show that the addition of cobalt to the rhodium clusters increases the local magnetic moments of the Rh atoms Mokkath et al. [29] studied the structural, electronic and magnetic properties of FenRhm (n + m ≤ 8) clusters using the gradient generalized approximation (GGA). The authors noted that the addition of the iron atoms to the rhodium clusters improve the magnetic properties of these last. In addition, the calculations show that the orbital d plays an important role in the magnetic properties, while the contribution of the s and p orbital is nearly negligible. Recently, the bimetallic RhxMny (x + y = 2–4) clusters have been studied using PBE/SDD method by Srivastava and al. [30]. Their obtained results indicate that Rh2Mn2 cluster possesses a higher stability than the other clusters, and the magnetic properties of RhxMny clusters are influenced strongly by the number of Rh and Mn atoms in the clusters. Using GGA-PW91 method, Lv et al. [31] studied the equilibrium geometries, electronic and magnetic properties of bimetallic ConRh (n = 1–8) clusters. Their results indicate that the Co2Rh, Co4Rh and Co7Rh clusters present a very big chemical stability than the other clusters. Besides, the calculated magnetic moments for these clusters is localized mainly on the cobalt atom. The same authors also studied the Structure, stability, and magnetism of (RhCo)n (n ≤ 5) clusters using the same method that described previously [32]. Their results indicate that the local magnetic moment of the cobalt atom increase after the addition of the rhodium atoms. This fact has been justified by the increasing Rh–Co bond in the clusters. The rhodium doping effect on the electronic properties of gold clusters has been studied by Yang et al. [33]. The obtained results show that the clusters with even number of atoms are more stable than those that have odd number of atoms. In addition, the Au5Rh cluster presents a very big chemical stability in comparison with the other clusters.
In this paper, we used the density functional theory (DFT) to study the structural, electronic and magnetic properties of the bimetallic RhnOs clusters. We also calculated the binding energy, fragmentation energy, second-order differences of energies, HOMO–LUMO energy gaps, vertical ionization potential, vertical electronic affinity, chemical hardness and chemical potential of these clusters. The obtained results will be analyzed and discussed.
Computational Details
In this work, the DFT method is used to calculate the geometrical structures optimizations and vibrational frequency analyses of RhnOs (n = 1–9) clusters provided by the Gaussian09 program [34]. The computational method is based on the M06-L functional [35]. The basis set has been used for this calculation including ECP triple-split type basis set termed as CEP-121G [36], which used previously for investigate the equilibrium structures and the spectroscopic properties of transition metal clusters [37–39].
In order to check the validity of the computational method for the prediction of the structural and physical properties of these clusters, we have compared different forms of exchange–correlation functional and basis sets with the experimental data for the small Os2 and Rh2 clusters. The calculated bond lengths, vibrational frequencies and binding energies together with available experimental data were reported in Table 1. For Os2 dimer, the calculated bond length was found to be 2.290 Å, which are in good agreement with the experimental data (2.314 and 2.27 Å) [40, 41]. The calculated binding energy is 1.390 eV/atom, which is smaller than the experimental value (2.15 eV/atom) [42]. In addition, our calculations indicate that the spin state of the lowest-energy Os2 dimer is septet confirming with Wu et al. and Du et al. [43, 44]. On the other hand, the spin state was found by Takahashi et al. [45] to be quintet. To our knowledge, there is no experimental values for the vibrational frequency to compare our theoretical result. Wu et al. [43] predicted a bond length of 2.283 Å, a binding energy value of 1.255 eV/atom and a vibrational frequency of 289.7 cm−1 using B3LYP/CEP-121G level. The bond length and the vibrational frequency were found by Du et al. [44] to be 2.281 Å and 282 cm−1, respectively, using BP86/CEP-121G level. Recently, Takahashi et al. [45] calculated the bond length to be 2.135 Å using PBE exchange–correlation. The ground state of Rh2 dimer is predicted to be quintet with a bond length of 2.264 Å, vibrational frequency of 311.04 cm−1, and binding energy of 1.466 eV/atom. Our theoretical results are not only in good agreement with the experiment [46, 47], but also better than the previous theoretical results. Reddy et al. [48] predicted a bond length and binding energy of 2.340 Å and 1.88 eV/atom, respectively, using a combination of molecular-dynamics and ab initio density-functional scheme including gradient corrections. Chien et al. and Xian et al. [49, 50] reported a bond length of 2.311 and 2.32 Å, and vibrational frequency of 282 and 289 cm−1, using the GGA method and PW91P86/Lanl2DZ level, respectively. Recently, Beltrán et al. [51] reported 2.27 Å as bond length and 0.8 eV/atom as binding energy, using rhe B3LYP/TZVP level. From our results, it has clearly to see that the M06-L/CEP-121G level is reliable and accurate enough to describe small RhnOs clusters.
To determine the lowest-energy structure of the RhnOs clusters, we have tested different initial geometries, linear, bi-dimensional and three-dimensional configurations at various possible spin multiplicities. In addition, harmonic vibrational frequencies calculations were conducted to confirm that the optimized structures correspond to a local minimum.
Results and Discussion
Geometrical Configurations
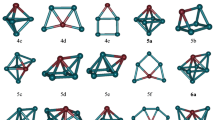
For each RhnOs cluster, we have optimized a big number of initial geometries. The lowest-energy structures and low-lying energy isomers are shown in Figs. 1 and 2. For these geometries, the spin multiplicity, symmetry, relative energy (ΔE) (relative to the lowest-energy structure), binding energy per atom (E b) and the shortest Rh–Rh and Rh–Os bond lengths are reported in Table 2.

The lowest energy and low-lying structures of RhnOs (n = 1–5) clusters. (na) The lowest energy structures. (nb–nf) Few low-lying structures

The lowest energy and low-lying structures of RhnOs (n = 6–9) clusters. (na) The lowest energy structures. (nb–nf) Few low-lying structures
The obtained results for RhOs dimer (1a) with C∞v symmetry indicates that the sextet spin state is lower in energy than all possible spin states. The calculated bond length is 2.277 Å, which lies between that of Rh2 (2.264 Å) and Os2 (2.290 Å) dimers. To our knowledge, no theoretical literature or experimental values available to us for RhOs dimer. The calculated binding energy is 1.666 eV/atom, which is larger than that of Rh2 dimer (1.466 eV/atom).
For Rh2Os cluster, the isosceles triangle (2a isomer) with C2v symmetry was found to be the most stable structure. The corresponding electronic state is 5A1. The calculated Rh–Os and Rh–Rh bond lengths are 2.315 and 2.734 Å. The binding energy was found to be 2.128 eV/atom. The other two low-lying isomers 2b and 2c are linear structures with different positions of the Os atom. The 2b isomer has D∞h symmetry with a binding energy value of 1.775 eV/atom. This configuration is 0.49 eV higher in energy than the 2a isomer, while the 2c structure with C∞v symmetry is higher than the lowest-energy structure by 1.74 eV.
In the case of Rh3Os cluster, the most stable structure (3a isomer) is an irregular triangular pyramid with Cs symmetry. The corresponding electronic state is 8A”. The shortest Rh–Os and Rh–Rh bond lengths are 2.446 and 2.607 Å, respectively. The 3b isomer is also a triangular pyramid with Cs symmetry is energetically higher than the equilibrium geometry (3a) by only 0.005 eV. Thus, both types of structures (3a and 3b) are almost degeneracy in energy. The next stable isomer (3c) is a rhombus structure with C2v symmetry. This is energetically higher than the ground state structure by 0.59 eV. For the last isomers (3d and 3e), which have the same symmetry (C2v). The difference of energy between these two isomers is 1.16 eV. The calculated binding energies of all the isomers are in the range of 2.013–2.560 eV/atom.
For Rh4Os cluster, we obtain a triangular bipyramid (the Os atom occupying the middle plane, 4a) as the lowest-energy structure with C2v symmetry. Its electronic state is 7A2. For this state, the binding energy per atom is 2.801 eV/atom to be 0.03 eV lower in energy than its similar structure (triangular bipyramid, where the Os atom localized on the apex, 4b). The square pyramidal structure (4c) with C4v symmetry is obtained as a metastable isomer. Its energy is only 0.04 eV higher than the energy of the most stable isomer (4a). The 4d isomer with Cs symmetry is obtained to be 1.07 eV higher in energy than the 4a isomer. For the 4e and 4f isomers, which are less stable than 4a isomer by 1.64 and 1.70 eV, respectively. The calculated binding energies of 4b, 4c, 4d, 4e and 4f are 2.794, 2.792, 2.586, 2.472 and 2.461 eV/atom, respectively.
The lowest-energy structure of Rh5Os cluster (5a) can be viewed as an octahedron structure (C4v symmetry) with Os atom at the apex. The corresponding electronic state is 10B1. The shortest Rh–Os and Rh–Rh bond lengths are 2.538 and 2.579 Å. The structure of the distorted prism (5b) with Cs symmetry is found to be 0.74 eV higher in energy than 5a isomer while a bicapped tetrahedral structure (5c) lies 1.05 eV than the equilibrium structure. For the 5d isomer (Cs symmetry), the calculated relative energy is higher than most stable structure by 1.21 eV. The last two isomers 5e (3D configuration) and 5f (2D configuration) are 1.41 and 2.69 less stable than the 5a isomer. The measured values of the binding energy for all the isomers of the Rh5Os cluster are in the range of 2.684–3.132 eV/atom.
For Rh6Os cluster, the 6a isomer with Cs symmetry has been reported to be the lowest-energy structure. Its electronic state and binding energy are 11A″ and 3.237 eV/atom, respectively. The second isomer (6b) with Cs symmetry is energetically higher than the 6a isomer by 0.30 eV. The calculated binding energy for this isomer is 3.194 eV/atom. The square capped prism structure (6c isomer) with C2v symmetry is obtained by adding one Os atom to the 5b structure. Its binding energy per atom is 3.151 eV/atom. The corresponding relative energy is 0.60 eV, which is less than the next two structures 6d (E b = 3.148 eV/atom) and 6e (E b = 3.107 eV/atom) by about 0.02 and 0.31 eV, respectively. The last ground state (6f) is a planar structure (C2v symmetry). It has 2.798 eV/atom of binding energy value to be the highest in energy for the Rh6Os cluster.
According to the calculated results on the Rh7Os cluster, all the obtained isomers are corresponding to octet spin state except the most stable isomer. This last isomer (7a) can be viewed as a bicapped octahedron structure with Cs symmetry and 8A′ electronic state. The calculated binding energy of this structure is 3.372 eV/atom. The shortest Rh–Os and Rh–Rh bond lengths are 2.474 and 2.561 Å, respectively. The second low-energy isomer (7b) is also a bicapped octahedron structure but the Os atom occupying the apex of the octahedron, while two Rh atoms were capped on the both sides of the Os atom. This configuration is just 0.16 eV higher in energy than the equilibrium geometry (7a). The third isomer (7c) can be considered as two prisms fused on a square face (Cs symmetry). The measured relative energy for this state is 0.77 eV higher than the low-lying isomer (7a). Another Cs type structure (7d isomer) has also 3D configuration, which is energetically 0.81 eV higher than the 7a isomer and 0.49 eV less than the 7e isomer (C2v symmetry). The 7f isomer (Cs symmetry) predicted as a sixth stable isomer (ΔE = 1.65 eV). The binding energies of 7b–f isomers are in the range of 3.165–3.351 eV/atom, respectively.
For Rh8Os cluster, the lowest-energy structures prefer the 3D configurations. The 8a isomer with C2v symmetry is energetically lower than the other 3D structures. The corresponding electronic state is 7A2. The calculated binding energy, Rh–Os and Rh–Rh bond lengths are 3.430 eV/atom, 2.483 and 2.588 Å, respectively. The next isomer (8b) with Cs symmetry is obtained to be 0.25 eV higher in energy than the 8a isomer. The calculated binding energy for this configuration is 3.403 eV/atom. The other three low-lying isomers (8c, 8d and 8e) are also 3D structures; their symmetries are C2, C2v and C4v. The isomer 8a is lower in energy by 0.79, 0.80 and 0.90 eV than the isomers 8c, 8d and 8e, respectively. The measured binding energies for these isomers are 3.343, 3.342 and 3.330 eV/atom, respectively.
Finally, we present the equilibrium geometries of Rh9Os cluster. The most stable structure can be considered as a combination between two octahedrons (9a) with Cs symmetry. The corresponding electronic state is 16B2. The calculated Rh–Os and Rh–Rh bond lengths are 2.551 and 2.590 Å, respectively. The pentagon prism (9b isomer) with Cs symmetry is energetically 0.88 eV higher than the most stable isomer. The 9c isomer with has also Cs symmetry with an energy of 0.91 eV higher than 9a isomer. The last two isomers (9d and 9e) are also 3D configurations with C2 and C4v symmetries, respectively. The energy difference between these two isomers is 1.58 eV. The calculated binding energies for all the isomers of the Rh9Os cluster are in the range of 3.301–3.590 eV/atom.
Relative Stability
In the aim to understand the relative stability of the ground-state RhnOs clusters, the binding energies per atom (E b), fragmentation energies (ΔE f ) and the second-order differences of energies (Δ2 E) of different cluster sizes have been calculated as follows:
where E (RhnOs), E (Rhn+1Os) et E (Rhn−1Os) represent the total energies of the ground-state structure of the RhnOs, Rhn+1Os and Rhn−1Os clusters, respectively. E (Rh) and E (Os) represent the total energy of the Rh and Os atoms.
In Fig. 3, we present the binding energy per atom of the RhnOs clusters. As we can see from this figure, the binding energy increases monotonically with the increasing cluster size, implying that these clusters can continuously gain energy during the growth process and the clusters become increasingly stabilized. The highest binding energy per atom value was observed for the Rh9Os cluster (3.590 eV/atom). In addition, the obtained results show that the binding energies per atom of these clusters are larger than that of pure Rhn+1 clusters (see Table 2), it indicates that the doping of the Os atom enhances the stabilities of the Rhn+1 clusters. In other words, the chemical reactivity of the RhnOs clusters can be improved after the doping.

Binding energy per atom for the lowest-energy structures of RhnOs clusters as a function of cluster size
The calculated values of the fragmentation energies (ΔE f ) are reported in Fig. 4. It’s clearly to see that ΔE f presents an odd–even oscillation as function as cluster size. Four peaks remarkable at n = 1, 3, 5 and 7, which indicate that the RhOs, Rh3Os, Rh5Os and Rh7Os clusters are more stable than their neighboring clusters.

Size dependence of the fragmentation energies (ΔE f ) of RhnOs clusters
The relative stability of these clusters can be also analyzed by examining the second-order differences of energies (Δ2 E). The variation of Δ2 E as function as cluster size is plotted in Fig. 5. The same remarkable peaks in the analysis based on the fragmentation energies at n = 1, 3, 5 and 7 for RhOs, Rh3Os, Rh5Os and Rh7Os clusters, to confirm that these clusters have a strong chemical stability.

Size dependence of the second-order energy difference (Δ2 E) of RhnOs clusters
HOMO–LUMO Gap
The HOMO–LUMO gap (highest occupied–lowest unoccupied molecular orbital gap) is an important parameter that characterizes the chemical stability of the clusters. In general, a large value of the HOMO–LUMO energy gap is related to an enhanced chemical stability, while a small one corresponds to a high chemical reactivity. The calculated HOMO–LUMO energy gaps for the most stable RhnOs clusters are reported in Table 3 and plotted in Fig. 6. As seen from Fig. 6, the local peaks are found at n = 1, 3, 5 and 7, indicating that the RhOs, Rh3Os, Rh5Os and Rh7Os clusters are more stable and have lower chemical reactivity than the other clusters. Moreover, the highest value of HOMO–LUMO energy gap was observed for the RhOs cluster (1.067 eV). The larger gap of the RhOs cluster should be mainly originate from his relative closure of electronic shell [53]. In addition, The HOMO–LUMO energy gap values are small, implying that these clusters have a metallic feature.

Size dependence of the HOMO–LUMO energy gaps of RhnOs clusters
Vertical Ionization Potential, Vertical Electronic Affinity, Chemical Hardness and Chemical Potential
In cluster physics, the vertical ionization potential (VIP) and the vertical electronic affinity (VEA) are used as important properties that reflect the size-dependent evolution of the electronic structure. In general, a higher value of VIP indicates a higher stability.
The Size dependence of the VIP is plotted in Fig. 7. As we can see, the VIP deceases rapidly until n = 6, then becomes nearly stable. The highest value of VIP is observed for the RhOs cluster (7.544 eV), indicating that this cluster is more stable than the other clusters. For the VEA, our results (see Table 3) indicate that the VEA increase rapidly with the cluster size. Moreover, we can easily see that the VEA values are much lower than the VIP values, indicating that these clusters can easily accept electrons.

Size dependence of the vertical ionization potential (VIP) of RhnOs clusters
From the calculations of VIP and VEA, we investigated the chemical hardness (η) and chemical potential (µ) as a function of cluster size. Chemical hardness is an electronic quantity that characterizes the relative stability of the clusters. A large value of the chemical hardness indicates that the cluster is less reactive [54]. η can also be seen as a resistance to charge transfer. On the other hand, µ describes the escaping of electrons from an equilibrium system. The chemical hardness and chemical potential can be expressed as follows:
The chemical hardness (η) and chemical potential (µ) values of the lowest-energy structures are summarized in Table 3 and plotted in Figs. 8 and 9. As shown in Fig. 8, η decreases rapidly with increasing cluster size. The higher value is observed for the RhOs cluster, indicating that this cluster is less reactive than the other clusters. This result is in excellent agreement with the previous analysis based on the VIP. For chemical potential (Fig. 9), the results exhibit that RhOs cluster has the smallest value of µ (−4.184 eV), indicating that RhOs cluster is the most stable one among all the clusters.

Size dependence of the chemical hardness for the lowest-energy structures of RhnOs clusters

Size dependence of the chemical potential for the lowest-energy structures of RhnOs clusters
From the analysis based on the fragmentation energies, second-order differences of energies, HOMO–LUMO energy gaps, VIP, chemical hardness and chemical potential, we can say that the RhOs cluster is the most stable among all the examined clusters. In other words, RhOs can be considered as a magic cluster.
Magnetic Properties
The total magnetic moment of RhnOs (n = 1–9) clusters has been calculated and the results are reported in Table 4. The variation of the magnetic moments for the lowest-energy structures as function as cluster size is shown in Fig. 10. As we can see, the magnetic moment of these clusters shows irregular oscillating behavior, and the magnetic moments oscillate with a maximum of 15 μ B for Rh9Os cluster and a minimum of 4 μ B for Rh2Os cluster. In addition, the results also exhibit that the magnetic moment of RhnOs clusters depend on their geometries and spin states (see Table 2). For example, the planar structures (3e) and (5e) have magnetic moment values of 1 μ B.

Total magnetic moment of RhnOs clusters as a function of cluster size
In order to investigate the local magnetic moments, we have performed the natural bond orbital analysis for the most stable RhnOs clusters. The local magnetic moment of 6s, 5d and 6p orbitals for the Os atom and 5s, 4d and 5p orbitals for the Rh atom are reported in Table 4. From this table, it is clearly seen that the total magnetic moment comes mostly from the magnetic moments of the Rh atoms for Rh3–9Os clusters. On the other hand, the contribution of the Os atom was observed for RhOs and Rh2Os clusters. In addition, the d orbitals play a dominant role in the determination of the magnetic behavior of these clusters. The s orbitals contribute little, while the contribution of the p orbitals is almost negligible.
Conclusions
In this work, we studied the geometric structures, relative stabilities, electronic and magnetic properties of the RhnOs clusters using DFT. The obtained results can be summarized as follows:
-
The geometric optimization shows that the three-dimensional structures are more stable than the bi-dimensional structures, and the atom Os always prefers the surface of the cluster.
-
From the results concern the binding energy, the energy of fragmentation, the second-order differences of energies and the HOMO–LUMO energy gaps, we found that the RhOs, Rh3Os, Rh5Os and Rh7Os clusters present a higher chemical stability than the other clusters.
-
The calculated values of VIP are much higher than the VEA values, implying that the RhnOs clusters can easily accept electrons.
-
The chemical hardness and chemical potential as function of cluster size were also investigated. The results indicate that the RhOs cluster has the lower chemical reactivity than other clusters. Furthermore, RhOs cluster is the most stable among all the examined clusters and it can be considered as a magic cluster. These results are in good agreement with the analysis based on HOMO–LUMO energy gaps, ΔE f , Δ2 E and VIP.
-
The calculations of the total magnetic moments of these clusters show that the magnetic moment come mostly from the Rh atoms for Rh3–9Os clusters, while the contribution of the Os atom was observed for RhOs and Rh2Os clusters. In addition, the d orbitals plays an important role in the magnetic moments of the RhnOs clusters.
References
G. Schmid (1992). Chem. Rev. 92, 1709.
L. N. Lewis (1993). Chem. Rev. 93, 2693.
X. S. Xu, S. Y. Yin, R. Moro, and W. A. de Heer (2005). Phys. Rev. Lett. 95, 237209.
E. K. Parks, T. D. Klots, and S. J. Riley (1990). J. Chem. Phys. 92, 3813.
W. P. Halperin (1998). Rev. Mod. Phys. 58, 533.
A. Soltani and A. Boudjahem (2014). Comput. Theor. Chem. 1047, 6.
T. Yonezawa, K. Imamura, and N. Kimizuka (2001). Langmuir. 17, 4701.
J. Y. Zhang, Q. Fang, A. J. Kenyon, and I. W. Boyd (2003). Appl. Surf. Sci. 208–209, 364.
C. D. Dong and X. G. Gong (2008). Phys. Rev. B 78, 020409.
T. Teranishi and M. Miyake (1998). Chem. Mater. 10, 594.
K. R. Gopidas, J. M. Whitesell, and M. A. Fox (2003). Nano. Lett. 3, 1757.
K. B. Sidhpuria, H. A. Patel, P. A. Parikh, P. Bahadur, H. C. Bajaj, and R. V. Jasra (2009). Appl. Clay. Sci. 42, 386.
A. Sanchez, M. Fang, A. Ahmed, and R. A. Sanchez-Dolgado (2014). Appl. Catal. A-Gen. 477, 117.
C. H. Campos, E. Rosenberg, J. L. Fierro, B. F. Urbano, B. L. Rivas, C. C. Torres, and P. Reyes (2015). Appl. Catal. A-Gen. 489, 280.
A. Behr, Y. Brunsch, and A. Lux (2012). Tetrahedron. Lett. 53, 2680.
A. J. Bruss, M. A. Gelesky, G. Machado, and J. Dupont (2006). J. Mol. Catal. A: Chem. 252, 212.
Y. Izumi, K. Konishi, M. Tsukahara, D. M. Obaid, and K. I. Aika (2007). J. Phys. Chem. C 111, 10073.
D. Han, X. Li, H. Zhang, Z. Liu, G. Hu, and C. Li (2008). J. Mol. Catal. A: Chem. 283, 15.
T. J. Yoon, J. I. Kim, and J. K. Lee (2003). Inorg. Chim. Acta. 345, 228.
A. J. Cox, J. G. Louderback, and L. A. Bloomfield (1993). Phys. Rev. Lett. 71, 923.
R. D. Adams and X. Qu (1995). Organometallics 14, 4167.
C.-T. Au, C.-F. Ng, and M.-S. Liao (1999). J. Catal. 185, 12.
T. Zoberbier, et al. (2012). J. Am. Chem. Soc. 134, 3073.
T. W. Chamberlain, T. Zoberbier, J. Biskupek, A. Botos, U. Kaiser, and A. N. Khlobystov (2012). Chem. Sci. 3, 1919.
F. M. Mendes and M. Schmal (1997). Appl. Catal. A-Gen. 163, 153.
A. Trunschke, H. Ewald, D. Gutschick, H. Miessner, M. Skupin, B. Walther, and H. C. Bottcher (1989). J. Mol. Catal. 56, 95.
X. Yang, D. Chen, S. Liao, H. Song, Y. Li, Z. Fu, and Y. Su (2012). J. Catal. 291, 36.
S. Dennler, J. Morillo, and G. M. Pastor (2003). Surf. Sci. 532–535, 334.
J. H. Mokkath and G. M. Pastor (2012). Phys. Rev. B 85, 054407.
A. K. Srivastava and N. Misra (2014). Comput. Theor. Chem. 1047, 1.
J. Lv, X. Bai, J. F. Jia, X. H. Xu, and H. S. Wu (2012). Physica B. 407, 14.
J. Lv, F. Q. Zhang, X. H. Xu, and H. S. Wu (2009). Chem. Phys. 363, 65.
J. X. Yang, C. F. Wei, and J. J. Guo (2010). Physica. B 405, 4892.
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian, Inc. Wallingford CT, 2009.
Y. Zhao and D. G. Truhlar (2006). J. Chem. Phys. 125, 194101.
W. J. Stevens, H. Basch, and M. Krauss (1984). J. Chem. Phys 81, 6026.
N. S. Venkataramanan (2008). J. Mol. Struct. Theochem 856, 9.
J. Du, H. Wang, and G. Jiang (2007). J. Mol. Struct. Theochem 817, 47.
W. Bouderbala, A. Boudjahem, and A. Soltani (2014). Mol. Phys. 112, 1789.
F. A. Cotton, A. R. Chakravarty, D. A. Tocher, and T. A. Stephenson (1984). Inorg. Chim. Acta. 87, 115.
C. D. Tait, J. M. Garner, J. P. Collman, A. P. Sattelberger, and W. H. Woodruff (1989). J. Am. Chem. Soc. 111, 9072.
M. D. Morse (1986). Chem. Rev. 86, 1049.
Z. Wu, B. Han, Z. Dai, and P. Jin (2005). Chem. Phys. Lett. 403, 367.
J. Du, X. Sun, and H. Wang (2008). Int. J. Quant. Chem. 108, 1505.
K. Takahashi, S. Isobe, and S. Ohnuki (2013). Chem. Phys. Lett 555, 26.
K. A. Gingerich and D. L. Cocke (1972). J. Chem. Soc. Chem. Commun. 1, 536.
H. Wang, H. Haouari, R. Craig, Y. Liu, J. R. Lombardi, and D. M. Lindsay (1997). J. Chem. Phys. 106, 2101.
B. V. Reddy, S. K. Nayak, S. N. Khanna, B. K. Rao, and P. Jena (1999). Phys. Rev. B 59, 5214.
C. H. Chien, E. Blaisten-Barojas, and M. R. Pederson (1998). Phys. Rev. A 58, 2196.
Y. J. Xian, W. Cheng-Fu, and G. Jian-Jun (2010). Physica. B 405, 4892.
M. R. Beltrán, F. B. Zamudio, V. Chauhan, P. Sen, H. Wang, Y. J. Ko, and K. Bowen (2013). Eur. Phys. J. D 67, 63.
A. Soltani, A. Boudjahem, and M. Bettahar (2015). Int. J. Quantum. Chem.. doi:10.1002/qua.25038.
M. X. Chen and X. H. Yan (2008). J. Chem. Phys. 128, 174305.
R. G. Parr and W. Yang Density Functional Theory of Atoms and Molecules (Oxford, New York, 1989).
Acknowledgments
The authors would like to acknowledge Pr. Abdaoui Mohammed (Director of Applied Chemistry Laboratory).
Author information
Authors and Affiliations
Corresponding author
Additional information
The article is being retracted at the request of the authors. This work in the article was initiated under the direction of and with the support of two senior scientists (Prof. A.-G.Boudjahem and Dr. M. Chettibi) who were consulted about and are not in concurrence with publishing this paper. The conclusions of the paper are preliminary as the research is not finalized.
About this article

Cite this article
Soltani, A., Bouderbala, W. & Boudjahem, Ag. RETRACTED ARTICLE: First Principles Study of the Geometries, Relative Stabilities and Magnetic Properties of Bimetallic RhnOs (n = 1–9) Clusters. J Clust Sci 27, 715–731 (2016). https://doi.org/10.1007/s10876-016-0970-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-016-0970-6