
Abstract
In this paper, we present theoretical investigations of electronic and magnetic properties of the ordered cubic ferromagnetic MnSe x S 1 −x , MnSe x Te1 −x , and MnSe x Po1 −x for x = 0.25, 0.50, and 0.75 using the full-potential linearized augmented plane wave method. Regarding structural properties, we found that the lattice parameters of MnSeS, MnSeTe, and MnSePo alloys follow Vegard’s law. Results show that MnSe x Te1 −x exhibits a half-metallic behaviour for all Mn concentrations, while MnSeS is half-metallic only for x = 0.5 and 0.75, and MnSePo is half-metallic only for x = 0.25. Moreover, a total magnetic moment of 5 µB is found for all alloys where the half-metallic phase is present. The total magnetic moment is found to be independent of composition and it is mainly due to the Mn atom.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
In the present years, there have been growing interest in the field of spintronics because it offers strong opportunities for the generation of multifunctional devices manipulating conventional charge-based microelectronics with the addition of spin degree of freedom [1]. Diluted magnetic semiconductors (DMS) [2] form a new class of materials, which present specific properties for the semiconductor spintronics field. The basic representatives of the DMS material class are II–VI semiconducting compounds alloyed with a Mn ion such as Cd1−x Mn x y (y = S, Se, Te) and exhibit many striking magnetic and magneto-optical properties originating from a hybridization between the Mn 3d and sp-band states [3–5].
The successful fabrication of heterostructures and multiple quantum well structures, which include DMS layers achieved with nonequilibrium growth techniques such as molecular-beam epitaxy (MBE), has significantly expanded II–VI investigations [6–8]. These techniques have also led to fabrication of compounds whose crystalline phases are different from equilibrium growth crystals. The previously hypothetical zinc blende (ZB) compounds of MnTe [9, 10], as well as MnS [11] and MnSe [12], have been grown by MBE. It is noted that in the case of MnS and MnSe, the stable crystal structure is of cubic NaCl type (α-MnSe and α-MnS) and both compounds are insulators. On the other hand, the stable phase of an equilibrium-grown MnTe crystal is a NiAs-type structure and is known as one of the few semiconductors among 3d transition metal compounds. It was also shown that below transition Néel temperature T N, the spin lattice associated with the Mn ions in ZB-Mn X VI (X = S, Se, Te) orders into a “type III” antiferromagnetic structure [10, 13–15]. Recently, ternary Cr-doped NiAs-type manganese tellurides, Mn1 −x Cr x Te, with x up to 14 %, have been fabricated, in which the substitution of Cr to Mn leads to a change from an antiferromagnetic (AFM) semiconductor of MnTe to a ferromagnetic (FM) (or ferrimagnetic) semiconductor of Mn1 −x Cr x Te [16]. This demonstrates the great interest in spintronics applications and justifies the study of magnetic materials based on zinc blende Mn X VI.
In this work, we perform a first-principles study on the structural, electronic, and magnetic properties of MnSe x S 1 −x , MnSe x Te1 −x , and MnSe x Po1 −x alloys in the ordered cubic ferromagnetic phase. The main goal here is to investigate the trends along the Mn X VI-doped Se series.
To the best of our knowledge based on literature survey, there is no theoretical work examining the various properties of Mn X VI (X = S, Te, Po)-doped Se. The paper is organized as follows. In “Section 2,” we briefly describe our method of calculation. “Section 3” contains the obtained results with discussions. The summary of the work will be given in “Section 4.”
2 Method
To perform the calculations, we use the full-potential linearized augmented plane wave (FLAPW) method within the density functional theory (DFT) [17], as implemented in the wien2k package [18]. The exchange and correlation effects are described in the generalized gradient approximation (GGA) [19], which is supposed to be more efficient for magnetic properties. In order to simulate the ordered cubic structures with x = 25 %, x = 50 %, and x = 75 % of Se for all compounds, we consider a model based on an eight-atom supercell, corresponding to a 1 × 1 × 1 conventional cubic cell. The basis function was expanded up to R MT K m a x = 8 (R MT is the plane wave radius and K m a x is the maximum modulus for the reciprocal lattice vector) for Mn X VI (X = S, Te, and Po) compounds and to 7.5 for ternary alloys. The maximum value for partial waves inside the atomic sphere is l = 10. Full relativistic approximation is used for core electrons, and scalar relativistic approximation is used for valence electrons. The muffin-tin radii for Se, Mn, and X = S, Se, and Te used in the FM calculations are listed in Table 1. Accurate Brillouin zone integration is performed using 3000 k-points for Mn X VI compounds and 200 k-points for the supercell calculations. Atomic relaxation is taken into account for all compositions. The iteration process is repeated until the calculated total energy of the crystal converges with less than 10−4 Ry and the maximum force on a single atom is less than 2.10−3 Ry/a.u.
3 Results and Discussions
In order to compute the ground-state properties, we calculate the total energies as a function of volume and fit them to the empirical Murnaghan equation of state [20]. The corresponding equilibrium lattice constants, bulk moduli, and cohesive energies of the ferromagnetic phase are given in Table 2 for both binary compounds and ternary alloys. No theoretical calculations or experimental data exist so far for MnSeX (X = S, Te, and Po); therefore, we will compare our results just with the parent elements MnTe and MnPo. Our calculations are in good agreement with those of Fang Zhu et al. [21] for MnTe and with those of Kahal et al. [22] for MnPo, whereas there is quite a large discrepancy between the calculated cohesive energy and the one reported by Wei and Zunger [23] for MnTe, considering that both have used the same method (LAPW). The reason is that in the present work, we use GGA formalism instead of LSDA like in the paper of Wei and Zunger [23].
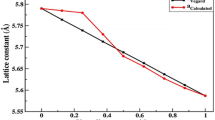
Figure 1 shows the calculated lattice constant as a function of selenium composition together with Vegard’s law variation assuming linearity [25]. The lattice parameters of MnSeS, MnSeTe, and MnSePo alloys deviate weakly from Vegard’s law.

a–c Calculated lattice parameters of MnSeX (X = S, Te, and Po) alloys versus composition compared to the virtual crystal approximation (VCA)
The spin-dependent total and partial density of states (DOS) of all ternary compounds at their equilibrium lattice constant for x = 0.25, 0.5, and 0.75 are depicted in Figs. 2, 3 and 4, respectively. All compounds are found half-metallic, except MnSeS for x = 0.25 and MnSePo for x = 0.5 and x = 0.75 since there are states available at the Fermi-level E f.

Total and partial spin-polarized density of states (DOS) of MnSe x S 1−x , MnSe x Te1−x , and MnSe x Po1−x alloys for x = 0.25

Total and partial spin-polarized density of states (DOS) of MnSe x S 1−x , MnSe x Te1−x , and MnSe x Po1−x alloys for x = 0.5

Total and partial spin-polarized density of states (DOS) of MnSe x S 1−x , MnSe x Te1−x , and MnSe x Po1−x alloys for x = 0.75
As shown by the partial density of state for all the MnSeX compounds (X = S, Te, and Po), the valence bands are constituted primarily from Mn d and (X, Se) p orbitals, which differ widely for spin-up and spin-down. For the majority spin, the valence band between −4.5 and −3 eV comes mainly from the Mn d states with a small contribution of (X, Se) p states. For the minority spin, the valence band between −6 and −3 eV comes from (X, Se) p states; while the conduction bands originate exclusively from the Mn d states.
In the tetrahedral crystal field, the Mn d state splits into t 2g and e g states, whereas the anion (Se or X atom) p sates have t 2p symmetry. The states with the same t 2 symmetry for spin-up (or spin-down) can overlap with each other forming bonding and antibonding states, while the e g states have a nonbonding character and do not take part in the hybridization process. This is clarified by the calculated spin-up charge density of MnSe x Te1 −x for x = 0.25 at the Γ point shown in Fig. 5. The lowest valence band for the majority spin at around −11.65 eV is essentially isolated anion (Se and Te) s states (Fig. 5a). The next band at around −4.44 eV is the bonding t 2g-p states reflecting the p-d bonding (i.e., the density lobes point toward the nearest neighbors) (Fig. 5b). The majority e g bands are lying in the valence bands at −3.41 eV. These bands reflect the nonbonding nature of the e g states as noticed from the charge density, which is highly localized on the Mn atoms, while there is a vanishing amplitude on the (Se, Te) atoms (see Fig. 5c). The next band at 0.064 eV shows the antibonding t 2g-p states (i.e., the density lobes point between the neighbor atoms, Fig. 5d). The band around 0.59 eV is mostly derived from (Se) s states (Fig. 5e).

Partial spin-up valence charge density of MnSeTe for x = 0.25 at Γ point in the (10) plane. a Energy at −11.65 eV, b energy at −4.44 eV, c energy at −3.41 eV, d energy at 0.064 eV, and e energy at 0.59 eV
Figures 6, 7 and 8 show respectively the spin-dependent DOS of the e g and t 2g levels of MnSeS, MnSeTe, and MnSePo. For the majority spin, the e g and t 2glevels are filled, while these states are empty for the minority spin. For all compounds, the levels are strongly Mn-localized (strong pick of the e g levels at −3.5 eV for the majority spin). This localization decreases for the MnSeX (X = S, Te, and Po) with increasing composition (i.e., x = 0.25 → 0.75).

Spin-polarized DOS of the e g and t 2g levels of MnSe x S 1−x for x = 0.25, 0.5, and 0.75

Spin-polarized DOS of the e g and t 2g levels of MnSe x Te1−x for x = 0.25, 0.5, and 0.75

Spin-polarized DOS of the e g and t 2g levels of MnSe x Po1−x for x = 0.25, 0.5, and 0.75
The calculated total and local magnetic moments for MnSeX alloys at the equilibrium lattice constants are given in Table 3. Calculated total integer magnetic moments are a typical character of half-metallic ferromagnets found by total DOS calculations. For all compounds, we found a global magnetic moment of 5 µB per Mn atom, except for MnSeS for x = 0.25 and MnSePo for x = 0.50 (0.75) where we found a total magnetic moment of 4.99 and 4.99 µB (4.98 µB). The total magnetic moments are mainly due to the Mn atoms, and the contribution from Se, Te, and Po is small. A relative contribution is noticed, however, from the interstitial region.
4 Summary
In summary, we have performed first-principles calculations based on the full-potential linearized augmented plane wave method to investigate the electronic and magnetic properties of MnSe x S 1−x , MnSe x Te1−x , and MnSe x Po1−x alloys for x = 0.25, 0.5, and 0.75 in their ordered cubic ferromagnetic phase. The results are summarized as follows:
-
a)
For the variation in the lattice parameter, we found for all alloys a weak deviation from Vegard’s law.
-
b)
We found that MnSe x Te1−x exhibits a half-metallic characteristic for all Mn concentrations. For MnSeS, the half-metallic character disappears for x = 0.25, while MnSePo is half-metallic only for x = 0.25.
-
c)
For MnSeTe, the calculated total magnetic moment gives 5 µB per Mn and remains constant with Mn composition, while MnSeTe and MnSePo show a total magnetic moment of 5 µB when these alloys are half-metallic.
References
Wolf, S.A., Awschalom, D.D., Buhrman, R.A., Daughton, J.M., von Molnar, S., Roukes, M.L., Chtchelkanova, A.Y., Treger, D.M.: Science 294, 1488 (2001)
Ohno, H.: Science 281, 951 (1988)
Taniguchi, M., Ley, L., Johnson, R.L., Ghijsen, J., Cardona, M.: Phys. Rev. B 31, 1206 (1986)
Ley, L., Taniguchi, M., Ghijsen, J., Johnson, R.L., Fujimori, A.: Phys. Rev. B 35, 2839 (1987)
Taniguchi, M., Fujimori, A., Fujisawa, M., Mori, T., Souma, I., Oka, Y.: Solid State Commun. 62, 431 (1987)
See articles in Diluted Magnetic Semiconductors, Furdyna, J.K., Kossut, J.: Semiconductors and semimetals, vol. 25. Academic, New York (1988)
Luo, H., Furdyna, J.K.: Semicond. Sci. Technol. 10, 1041 (1995)
Ramdas, A.K., Rodriguez, S. In: Cardona, M., Guntherodt, G. (eds.) : Light scattering in solids VI, p 137. Springer, New York (1991)
Durbin, S.M, Han, J., Sungki, O., Kobayashi, M., Menke, D.R., Gunshor, R.L., Fu, Q., Pelekanos, N., Nurmikko, A.V., Li, D., Gonsalves, J., Otsuka, N.: Appl.Phys. Lett. 55, 2087 (1989)
Klosowski, P., Giebultowicz, T.M., Rhyne, J.J., Samarth, N., Luo, H., Furdyna, J.K.: J. Appl. Phys. 70, 6221 (1991)
Okajima, M., Tohda, T.: J. Cryst. Growth 117, 810 (1992)
Kolodziejski, L.A., Gunshor, R.L., Otsuka, N., Gu, B.P., Hefetz, Y., Nurmikko, A.V.: Appl. Phys. Lett. 48, 1482 (1986)
Samarth, N., Kłosowski, P., Luo, H., Giebułtowicz, T.M., Furdyna, J.K., Rhyne, J.J., Larson, B.E., Otsuka, N.: Phys. Rev. B 44, 4701 (1991)
Ando, K., Takahashi, K., Okuda, T., Umehara, M.: Phys. Rev. B 46, 12289 (1992)
Hastings, J.M., Corliss, L.M., Kunnmann, W., Mukamel, D.: Phys. Rev. B 24, 1388 (1981)
Li, Y.B., Zhang, Y.Q., Sun, N.K., Zhang, Q., Li, D., Li, J., Zhang, Z.D.: Phys. Rev. B 72, 193–308 (2005)
Hohenberg, P., Kohn, W.: Phys. Rev. 136, B864 (1964)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., wien2k, J. Luitz.: an augmented plane wave program for calculating crystal properties. Vienna University of Technology, Vienna, Austria (2001)
Perdew, J.P., Burke, K., Ernzerhof, M.: Phys. Rev. Lett. 77, 3865 (1996)
Murnaghan, F.D.: Proc. Natl. Acad. Sci. U.S.A. 30, 244 (1944)
Zhu, Li-Fang, Liu, Bang-Gui: Phys. Status Solidi B 246, 1094 (2009)
Kahal, L., Zaoui, A., Ferhat, M.: J. Appl. Phys. 105, 063–905 (2009)
Wei, S.H., Zunger, A.: Phys. Rev. B 35, 2340 (1987)
Balzarotti, A., Czyzyk, M., Kisiel, A., Motta, N., Podgorny, M., Zimnal-Starnawska, M.: Phys. Rev. B 30, 2295 (1984)
Vegard, L.: Z. Phys. 5, 17 (1921)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Adli, W., Zaoui, A. & Ferhat, M. First-Principles Calculations of Electronic and Magnetic Properties in Ferromagnetic MnSeS, MnSeTe, and MnSePo Ternary Systems. J Supercond Nov Magn 29, 839–845 (2016). https://doi.org/10.1007/s10948-015-3335-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-015-3335-8