
Abstract
Mechanically robust silica xerogel/alumina fiber composites were successfully prepared by a cost-effective ambient pressure drying route. Microstructure investigation shows that the alumina fibers embed into the silica xerogel matrix as reinforcements, which improves the strength of the composites greatly. As the alumina fibers volume increases to 4.0%, the compressive and flexural strength value of the composite is 0.96 MPa and 2.53 MPa, while the thermal conductivity at room temperature slightly changes between 0.051 and 0.059 W/mK. The composite is hydrophobic and keeps water repellent until 300 °C as shown by the TG–DSC result. After being heated at 800 °C for 25 min, the composite remains structurally intact demonstrated good thermal resistant property. The above characteristics indicate that the as prepared composite has great potential application as thermal insulation material in energy-saving area.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Nowadays, energy conversation is becoming more and more important for sustainable development of human society. A variety of fields such as buildings, petrochemical, metallurgy and furnaces, etc. waste a large quantity of energy through heat loss. High performance thermal insulation material is recognized as one of the most effective measures being adopted to meet the demands of improving the efficiency of above mentioned fields [1,2,3,4,5]. In the past decade, silica aerogels with high porosity of more than 90% and ultra-low thermal conductivity of 0.02 W/mK under room temperature have attracted considerable attention as an emerging superinsulator all over the world. Many aerogel-based insulation materials are widely studied as an alternative to the conventional inferior insulators [6,7,8]. However, the expanded application of silica aerogels has been prevented seriously by the poor mechanical property and high production cost. To overcome the fragility limitations and reduce the overall price of the silica aerogels, many efforts have been directed toward the development of mechanically more robust aerogels with cost-effective procedures [9, 10].
The most important approach adopted to strengthen the silica aerogel is by incorporation of various fibers as reinforcement [11,12,13,14]. Composites that contain fibers and aerogel matrix are obtained by impregnating different fiber batting using silica sol and drying in supercritical fluids. Aspen Aerogels Inc. has successfully produced the fiber-reinforced aerogel blanket with improved thermal insulation and mechanical performances [15]. Although aerogel blankets possess good insulation properties, the aerogel tends to flake off and shed from the composite over time. Moreover, these aerogel blankets are flexible, which limited their applications where rigid insulation materials are required. Another disadvantage is that aerogels are dried by supercritical media in order to inhibit shrinkage and maintain highly porous nanostructure. However, supercritical drying process needs special autoclave and operates under high pressure and temperature, which is dangerous. Furthermore, supercritical fluid procedure is the major obstacle to reduce the preparation cost of various aerogel materials.
Due to the inherent shortcomings of the supercritical process, ambient pressure drying has been more extensively investigated in recent years [9]. Generally, wet gels are decorated by hydrophobic silylation agents and then evaporate the liquid in the pore under ambient pressure. This process is more safe and cost-effective than the supercritical drying route, which significantly promotes the economical production of aerogels in large scale and commercial applications. Under ambient drying conditions, the so-called xerogel is normally obtained. Therefore, the main motivation of this work is to develop a cost-effective rigid silica xerogel/alumina fiber composite and explore its mechanical and thermal insulation properties.
2 Experimental procedure
2.1 Materials
The chemical reagents used for the preparation of rigid silica xerogel/alumina fiber composites were tetraethylorthosilicate (TEOS), ethanol (EtOH), hydrochloric acid (HCl, 36 wt%), ammonium hydroxide (NH4OH, 27 wt%), hexamethyldisilazane (HMDS) and hexane. All these reagents were analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd (SCRC, China) without further purification. Alumina blanket (Saffil®, Unifrax, USA) with density of 96 kg/m3 was adopted as reinforcement of the silica xerogel/alumina fiber composites.
2.2 Preparation of the silica xerogel/alumina fiber composite
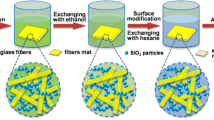
The preparation process of the rigid silica xerogel/alumina fiber composite is shown in Fig. 1. Firstly, silica sol was prepared according to a conventional two-step sol–gel method [16]. Typically, TEOS, EtOH, H2O and HCl were mixed and stirred at 55 °C for more than 2 h for complete hydrolysis. The obtained solution was cooled to room temperature and then NH4OH was added and stirred vigorously for 30 min to get the silica sol. The molar ratio of the TEOS, EtOH, H2O, HCl and NH4OH is 1:4:4:1.8 × 10−3:3.6 × 10−3. Secondly, the obtained silica sol was poured into the alumina fiber mat, which is fixed between two boards, and gelation occurred after about 2 h. The silica gel/alumina fiber composite was aged in ethanol for 24 h. Thirdly, the aged silica gel/alumina fiber composite was immersed into hexane to exchange the confined ethanol in the pores. This procedure was done two times within 48 h. Then the silica gel/alumina fiber composite was transferred into HMDS/hexane solution (v/v = 1:10) to modify the surface of the silica gel for 24 h. Finally, the modified silica gel/alumina fiber composite was drying under ambient pressure at 100 °C for 8 h and the rigid silica xerogel/alumina fiber composite was obtained.

Scheme for the preparation of the silica xerogel/alumina fiber composite
2.3 Characterization
The bulk densities of the silica xerogel/alumina fiber composites were determined by measuring the dimensions and mass of each composite sample. The microstructure of the composite was observed under scanning electron microscopy (SEM, Apollo300, CamScan and S4800, Hitachi, Japan). N2 adsorption–desorption isotherm of the silica xerogel was obtained on a Quadrasorb I analyzer (Quantrom, USA). Before the measurements, the samples were degassed in vacuum at 120 °C for 12 h. Pore size distribution of the silica xerogel was calculated from the desorption branch of the isotherm. The FT-IR spectrum was recorded by Spectrum GX (PerkinElmer, USA) in the region of 4000–400 cm−1 using the KBr method to investigate the surface medication of the silica xerogel matrix. The static contact angles of the composite samples were determined from the water drop shape placed on the sample surface to certify the hydrophobic surface (OCA20, Dataphysics, Germany). The thermal stability analysis was tested using TG–DSC (SDT-Q600, TA, USA) with a heating rate of 10 °C/min from room temperature to 600 °C in air. The uniaxial compression tests and three-point bending tests of the silica aerogel/alumina fiber composites were carried out on the universal testing machine (CMT5105, MTS, China). The sample dimension for the compressive strength was 20 mm × 20 mm × 15 mm and the loading rate was 2 mm/min. The tested samples for flexural strength had a dimension of 80 mm × 10 mm × 6 mm and the loading rate was 2 mm/min until material failure. Thermal conductivities of the silica xerogel/alumina fiber composites at room temperature were measured by HC-074 thermal conductivity tester (EKO Instruments, Japan) according to the ASTM C518. Briefly, the composite sample with dimension of 300 mm × 300 mm × 15 mm was placed between two plates in the test stack and a temperature gradient is established over the thickness of the material. The instrument electronics detect equilibrium according to the defined conditions and a final thermal conductivity value was collected. Thermal insulation property at 800 °C was tested on a molybdenum silicide bar electric furnace heating system, which is able to heat one side of the sample to 800 °C and recorded the temperature response of the opposite side of the sample. The detailed information about the single-sided heating equipment is referred to the published document [17].
3 Results and discussion
Figure 2 presents the SEM and photo pictures of the silica xerogel/alumina fiber composites. It is seen that the alumina fibers distribute randomly in-plane direction and the silica xerogel fill the cavities between the fibers. The silica xerogel matrix is not monolithic and crumbles into many blocks since the constrained shrinkage imposed by the fibers cause the formation of the cracks during drying. The size of the broken silica xerogel pieces are less than 100 µm. It is worth noting that almost all the cracked monolith of the silica xerogel adhere to the alumina fibers as shown in Fig. 2d. The bonding between the silica xerogel and the alumina fiber is of great importance for avoiding dust during handling of the composite. Figure 2e shows the high magnification image of the silica xerogel matrix and it is found that the xerogel network are composed of uniform secondary silica particles with diameter of about 30 nm. Large pieces of composite boards were cut to examine the machinery performance of the composites and the results indicate that the composites are strong enough to be cut into any expected shapes. The improvement of the mechanical performance of the composites arises from both the alumina fiber reinforcement and the silica xerogel matrix. Though many research have focused on obtaining xerogels with as low shrinkage as possible, we tried to prepare silica xerogel with a little higher density by using less surface modification reagent and avoid the disadvantage of the brittleness of the xerogel to some extent. Moreover, less hydrophobic reagent is also environmental friendly for the preparation of the composites.

SEM and photo pictures of the silica xerogel/alumina fiber composites with different densities: a 0.41 g/cm3, b, d 0.38 g/cm3, c 0.36 g/cm3, e silica xerogel matrix and f machined composite boards
Figure 3 presents the N2 adsorption–desorption isotherm and pore size distribution of the silica xerogel matrix. The sorption isotherm of the xerogel matrix exhibits the type IV morphology, which indicates the presence of mesopores between silica aggregates. The characteristic feature of the hysteresis is attributed to the existence of the so-called ink-bottle like cavities and the steepness of the desorption branch indicates the uniformity in diameter of the pores. The pore diameter distribution is very narrow and the most probable pore size is centered at 4 nm. Furthermore, micropores existed within the silica aggregates are confirmed from the pore size distribution curve of the silica xerogel. Both the meso- and microporous structure are relative to the contract of the silica xerogel backbone. Since the added water in the acid catalyzed step is under stoichiometric, the water is rapidly consumed to produce a distribution of partially hydrolyzed monomers which subsequently condense to form low molecular weight oligomeric species. When the base catalysts are added in the second step, the remaining alkoxide sites hydrolyze, which render all sites approximately equally reactive. So the condensation is primary between the oligomeric species and lead to a high branched structure of the silica network. With the removal of the solvent, the condensed oligomeric species shrink and rearrange. Therefore, the skeleton of the ambient pressure dried silica xerogel is characterized by a globular structure. Mesopores exist between the aggregates and micropores occur within the aggregate itself. The narrow pore size distribution indicates that dimension of the silica clusters are uniform, which is agree well with the SEM results. The cross-linked structure of the silica nanoparticles also suggests the contraction process is well controlled in the reaction conditions. This nanoporous structure is beneficial to the thermal insulation performance of the composites, due to the restriction of the gaseous thermal conductivity.

N2 adsorption–desorption isotherm of the silica xerogel matrix
It is believed that the shrinkage of the silica xerogels is related to the hydrophobic surface. FTIR spectrum of the silica xerogel matrix is depicted in Fig. 4. The vibration signals around 1088 cm−1 are typical Si–O–Si bands attributed to the condensed silica network. The peaks around 1632 cm−1 and the broad absorption around 3437 cm−1 are due to water and Si–OH groups, respectively. The Si–C peaks at 1257 and 849 cm−1 are due to the Si–(CH3)3 groups introduced by the HMDS modifier which contain three non-hydrolysable –CH3 groups [18, 19]. The bands around 2965 cm−1 assigned to C–H stretching vibration confirm the presence of methyl groups on the surface of the silica xerogel. The appearance of both Si–C and C–H absorption peak suggests that there are abundant non-hydrolysable organic substituent groups attached to the gel skeleton during the surface modification. These organic substituent groups endow with hydrophobicity of the silica xerogel/alumina fiber composites.

FTIR spectrum of the silica xerogel
Hydrophobic behavior of the silica xerogel/alumina fiber composites were also evaluated by studying their water contact angle values. Figure 5 shows water droplet on the composite surfaces with contact angle of 135°, 137° and 138° respectively. The high contact angles indicate that the hydrophobic character of the silica xerogel/alumina fiber composites. It is believed that the HMDS provides the hydrophobic groups (Si–CH3) by replacement of H from the Si–OH during the surface modification process.

Contact angles for the silica xerogel/alumina fiber composites with different densities: a 0.36 g/cm3, b 0.38 g/cm3 and c 0.41 g/cm3
Thermal stability of the surface pended organic groups of the silica xerogel/alumina fiber composites were analyzed by thermogravimetry and differential scanning calorimetry. Figure 6 shows the TG–DSC curves of the silica xerogel/alumina fiber composite with density of 0.38 g/cm3. There are two stages of weight loss during the whole heating process. The first stage from room temperature to about 300 °C is attributed to the evaporation of water and the remained organic solvent molecular adsorbed in the composite. The considerable weight loss in the second stage between 300 and 500 °C is ascribed to the oxidation of the Si–(CH3)3 groups on the surface of the silica xerogel matrix, which lead to the noticeable exothermic peak at 367 °C [20]. The results show that the retention of hydrophobicity of the composites is up to 300 °C and above this temperature the xerogel may become hydrophilic.

TG–DSC of the silica xerogel/alumina fiber composite
As is known to all, native silica xerogel are generally fragile and break at very small compressive strains. However, the compressive stress–strain curves in Fig. 7 suggest that the silica xerogel/alumina fiber composites are tough and show good fracture resistance. The compression curves consist of three regions: the first linear stage, the elastic–plastic stage and the second linear stage, which are related to the elastic deformation, compaction, and densification of the composites experienced during compression. The elastic deformation in the first stage for strains < 2% is attributed to the silica xerogel matrix. The specimen with density of 0.36 g/cm3 exhibits the least stress, which may be due to the delamination between the layers the alumina blanket during the silica xerogel drying. The elastic–plastic region is followed by a gradual increase in the strain range of 2–8%. This is probably attributable to the plastic deformation and rupture of the xerogel matrix. After the second region, a steep linear increase in the stress is observed at strain > 8%. This is attributable to elimination of pores through densification of the composites. At this stage, the stress rises rapidly with the increasing strain, during which the xerogel matrix and fibers resist the compressive stress together.

Compressive stress–strain curves of the silica xerogel/alumina fiber composites
The flexural strength of the silica xerogel/alumina fiber composites have been considerably improved due to the reinforcement of the alumina fiber blanket. As shown in Fig. 8a, all the flexural stress–strain curves exhibit linear increase until reaching the maximum strain with flexural strength of 1.26 MPa, 1.82 MPa and 2.53 MPa respectively, whereas the slope of the linear relationship reduces with decreasing density of the composite. Because the alumina fibers of the blanket orient in the plane direction, composite with high density would contain more layers of fiber than low density sample and result in better flexural performance. The decline after the apex of the flexural stress–strain curves suggests the final fracture of the tested specimen.

Flexural stress–strain curves (a) and flexural modulus (b) of the silica xerogel/alumina fiber composites
The flexural strength corresponding to the highest point of the flexural stress–strain curves of the composites are shown in Fig. 8b. Meanwhile, the flexural modulus of the silica xerogel/alumina fiber composites are also shown in Fig. 8b. Both flexural strength and flexural modulus show approximately linear increase with the growth of density of the composite. The flexural strains of the failure points are < 15% for all the samples, which also indicate the excellent mechanical property of the composite. The volume content of the alumina fiber plays a key role in controlling the mechanical behavior of the composites, as the reinforced fibers are much stronger than the silica xerogel matrix (Table 1).
Thermal conductivities of the silica xerogel/alumina fiber composites under room temperature are shown in Fig. 9. The composite with density of 0.38 g/cm3 possess the lowest thermal conductivity of 0.051 W/mK. The thermal conductivity of the composite with density of 0.41 g/cm3 is 0.059 W/mK, and the increase of the thermal conductivity may be attributed to the increased contribution of the solid phase. However, composite with density of 0.36 g/cm3 also exhibits an obvious increase of thermal conductivity to 0.058 W/mK. That may be due to the formation of cracks between the layers of the alumina fiber blanket and lead to the increase of the gaseous thermal conductivity [21].

Thermal conductivity of the silica xerogel/alumina fiber composite
Figure 10 presents the insulation performance of the composite with density of 0.38 g/cm3 at 800 °C. When one side of the material is heated to 800 °C, the temperature response recorded on the cold side show slow rise before 250 s, steep increase between 250 and 800 s, and reach equilibrium after 1000 s. The temperature at 1500 s is 670 °C. The noticeable drop of the temperature within the thickness of 8 mm indicates the excellent heat insulation property of the composite. It is reasonable to infer that larger temperature gradient will observed if thicker material was used in the test. Furthermore, the specimen kept intact and no cracks or delamination phenomenon were observed after the high temperature insulation experiment, which indicates that the composite is incombustible and has good structural stability under high temperature.

Thermal insulation performance of the silica xerogel/alumina fiber (sample dimension: 150 mm × 150 mm × 8 mm, ρ = 0.38 g/cm3)
4 Conclusions
In summary, rigid silica xerogel/alumina fiber composites with significantly improved strength have been fabricated by the cost-effective ambient pressure drying method. The more alumina fibers are incorporated, the stronger composite is obtained. The improved mechanical property is beneficial to the composite to be mechanized into any shapes for practical applications. The thermal conductivity at room temperature is 0.051 W/mK when the alumina fiber content is 3.5% by volume. Moreover, the as prepared composites are thermal and structurally stable under 800 °C. These characteristics indicate that the silica xerogel/alumina fiber composites have great application prospects in thermal insulation fields.
References
M. Talou, P. Miranzo, M. Camerucci, Int. J. Appl. Ceram. Technol. 14, 738 (2017)
R. Baetens, B.P. Jelle, A. Gustavsen, Energy Build. 43, 761 (2011)
R. Zhang, C. Ye, B. Wang, J. Porous Mater. 25, 171 (2018)
T. Shimizu, K. Matsuura, H. Furue, K. Matsuzak, J. Eur. Ceram. Soc. 33, 3429 (2013)
R. Zhang, X. Hou, C. Ye, B. Wang, J. Alloys Compd. 699, 511 (2017)
M. Koebel, R. Arnaud, P. Achard, J. Sol-Gel Sci. Technol. 63, 315 (2012)
J. Laskowski, B. Milow, L. Ratke, J. Non-Cryst. Solids 441, 42 (2016)
M. Reim, J. Manara, S. Korder, M. Arduini-Schuster, H.-P. Ebert, J. Fricke, Sol. Energy 79, 131 (2005)
M. Koebel, L. Huber, S. Zhao, W.J. Malfait, J. Sol-Gel Sci. Technol. 79, 308 (2016)
P.R. Aravind, P. Shajesh, G.D. Soraru, K.G.K. Warrier, J. Sol-Gel Sci. Technol. 54, 105 (2010)
J. He, X. Li, D. Su, H. Ji, X. Wang, J. Eur. Ceram. Soc. 36, 1487 (2016)
Z. Li, X. Cheng, S. He, X. Shi, L. Gong, H. Zhang, Compos. A 84, 316 (2016)
M. Li, H. Jiang, D. Xu, J. Porous Mater. 25, 1417 (2018)
J. He, X. Li, D. Su, H. Ji, Y. Qiao, J. Mater. Sci. 50, 7488 (2015)
A. Hoseini, C. McCague, M. Andisheh-Tadbir, M. Bahrami, Int. J. Heat Mass Transfer 93, 1124 (2016)
T.M. Tillotson, L.W. Hrubesh, J. Non-Cryst. Solids 145, 44 (1992)
L. Shang, D. Wu, Y. Pu, H. Wang, F. Wang, Z. Gao, Ceram. Int. 42, 3351 (2016)
W.J. Malfait, S. Zhao, R. Verel, S. Iswar, D. Rentsch, R. Fener, Y. Zhang, B. Milow, M. Koebel, Chem. Mater. 27, 6737 (2015)
V.G. Parale, D.B. Mahadik, S.A. Mahadik, M.S. Kavale, R.A. Venkateswara, P.B. Wagh, J. Sol-Gel Sci. Technol. 63, 573 (2012)
J.L. Gurav, A.V. Rao, Y. Nadargi, J. Sol-Gel Sci. Technol. 50, 275 (2009)
G. Lu, X. Wang, Y. Duan, X. Li, J. Non-Cryst. Solids 357, 3822 (2011)
Acknowledgements
This study was funded by the National Natural Science Foundation of China (Grant Number 51576058). The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, J., Wu, W., Yang, H. et al. Rigid silica xerogel/alumina fiber composites and their thermal insulation properties. J Porous Mater 26, 1177–1184 (2019). https://doi.org/10.1007/s10934-018-0711-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-018-0711-3