
Abstract
Magnetic mesoporous nanocomposites are emerging as a significant new material to improve the effectiveness of cancer treatment and enhance the availability of drug therapy. Thus, the core–shell structure magnetic mesoporous nanocomposites (Fe3O4@SiO2@LDH) were synthesized with an average diameter of about 100 nm and used as methotrexate (MTX) carriers for cancer therapy. As an important biological material, Fe3O4@SiO2@LDH exhibited higher superparamagnetic behavior and biocompatibility, and the drugs can be loaded in the channels of the mesoporous silica and the interlayer of layered double hydroxides (LDH). The embedding rate of magnetic mesoporous silica (Fe3O4@SiO2) and LDH were 60.32 % and 0.67 %, respectively, and the release rate of the drug delivery systems (Fe3O4@SiO2@LDH-MTX) was 66.81 %. In addition, LDH as a kind of pH-sensitive material was adopted for controlled drug release and WST-1 assays in cancer cells (Hela) demonstrated that Fe3O4@SiO2@LDH-MTX presented high anti-tumor activity, while the carriers were nearly non-toxic. Therefore, all the results suggested that the magnetic nanocomposites can be employed to deliver MTX, and would be applied in the field of cancer therapy in the future.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
In recent years, with the rapid development of nano science and technology, a wide variety of functional nanomaterials were prepared and used in catalytic science [1], biological medicine [2] and photonics [3], especially silica-based nanostructured mesoporous materials, which have been the hot-point in the research of material preparation [4]. Nano mesoporous silica has excellent bioaffinity, ordered pore network,large pore volume and a relatively high surface area, so it is suitable as pharmaceutical carriers [5], and many scholars used them to transport small molecules [6], biological agents [7], proteins [8] or DNA [9] to achieve the purpose of targeted delivery in the field of molecular imaging and target identification. In addition, with the deepening of research, magnetic mesoporous silica [10], a new kind of nanocomposite material, has showed a greater application prospect for improving the transport efficiency of drug carriers, which is mainly attributed to its excellent magnetic targeting property.
Magnetic mesoporous silica nanocomposites applied in the field of biological drug delivery has the complete core–shell structure [11]. Firstly, their nucleus are the magnetite with high superparamagnetic, under external magnetic field, nanoparticles can be humanly transported to the target sites [12]. Therefore, the availability of drugs will also have an obvious improvement. Furthermore, the outer layer of the nanocomposites wrapped in mesoporous silica with short pore channels. These channels are the large amount of available pore volume, which can store drugs, proteins, nucleic acids or other molecules [13]. What’s the most important point is that the mesoporous silica with adjustable pore size can be attained through changing the length of surfactants’ carbon chain, this can greatly increase the loading rate. Besides, magnetic mesoporous silica composites can also keep the particle size below 100 nm, ensuring the controllable size of the materials is small enough to evade enhanced permeability and retention (EPR) of the body [14]. Finally, although drugs can be efficiently stored in the channels of the mesoporous silica, how to control the drug release effectively is still a very important task, it is because the anti-cancer drugs exist a certain degree of damage to normal cells [15]. Several studies have indicated that the pH value of most solid tumors (pH < 6.0) is lower than the surrounding normal tissues (pH 7.4) [16]. Therefore, using the pH-sensitive LDH as a switch to protect the drug or control drug release plays an important role in drug controlled release system.
At present, on account of the significant advantages of magnetic mesoporous silica, using it as a biological carrier material has been more and more common. Zhang et al. [17] prepared magnetic mesoporous silica with a particle size of about 100 nm, and 1,3,5-trimethylbenzene was selected as expanding agent to study the influences of the mass ratio of 1,3,5-trimethylbenzene to cetyltrimethylammonium bromide (CTAB) on the pore size and morphology of the composite nanoparticles. Chen et al. [18] prepared ellipsoidal magnetic silica and then a new layer of mesoporous silica was coated on the surface of silica to deliver doxorubicin for the treatment of cancer. Besides, some academics used nanoparticles as valves to control drug release. Slowing et al. [19] prepared ordered mesoporous silica and put the drugs in the pipelines of mesoporous silica, then modified CdS nanoparticle sealed the both ends of the channel. Moreover, by wrapping the drug carriers with DNA fragments, the gene can also be delivered to target sites.
LDH, a type of pH-sensitive layered material, will be destroyed in acidic conditions [20], which can show such an advantage in controlling drug release. Additionally, LDH also plays a role in drug loading, because a small amount of anionic drugs can be loaded in the interlayer of LDH [21], even if they are different from the drugs loaded in the channels of mesoporous silica [22]. In general, under acidic environment, the plate layers open and the drug is released. Moreover, the LDH coated on the surface of the silica will be broken and drugs in the pipelines of mesoporous silica will also be released. Basic on this, in this article, we prepared a small particle size of magnetic mesoporous silica nanoparticles by the method of mixing, and MTX was housed in the channels of mesoporous silica. Finally, we coated a thin LDH layer on the surface of the carriers to load a small amount of extra MTX and control the release of MTX (Fig. 1). Through a series of tests, it was found that the nanoparticles can be used to deliver MTX for cancer therapy.

The description of experimental scheme
2 Materials and methods
2.1 Materials
Ferric chloride (FeCl3·6H2O), ferrous chloride (FeCl2·4H2O), ethanol, nitric acid (65–68 wt%) ammonia hydroxide (25 wt%), cetyltrimethylammonium bromide (CTAB), ethyl orthosilicate (TEOS), ammonium nitrate (NH4NO3) and magnesium nitrate (Mg(NO3)2·6H2O) were supplied by Sinopharm Chemical Reagent Co., Ltd., Shanghai, China., oleic acid, chloroform, isopropanol, ethylene glycol and aluminium isopropoxide were supplied by Shanghai Lingfeng Chemical Reagent Co., Ltd., Shanghai, China. 3-aminopropyltriethoxysilane (APTES) and aluminium isopropoxide were purchased from Aladdin. Methotrexate (MTX) was purchased from Yuanye Biotechnology Co., Ltd., Shanghai, China. 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST-1) and Hela were supplied by Beyotime Biotechnology Co., Ltd., Shanghai, China. All chemicals and solvents used in this study were analytical grade and without further purification.
2.2 Characterizations
Fourier-transform infrared spectroscopy (FT-IR) spectra were detected on Vector 22 spectrometer (Bruker Co., Ltd., Germany). The crystalline phases of the samples were identified by X-ray diffraction (XRD, Bruker Co., Ltd., Germany) with Cu Kα radiation. Magnetic properties of the nanoparticles were recorded on vibrating sample magnetometer (VSM, Model 7410, Lake Shore Co., Ltd., USA). The porous structure, the surface area, pore volume and pore diameter were detected by BET analysis instrument (Quantachrome Co., Ltd., America). The size, morphology and energy spectrum analysis of the samples were investigated on transmission electron microscopy (TEM, Model Tecnai 12, Philips Co., Ltd., Holland) and field emission scanning electron microscopy (FESEM, Model S4800II, Hitachi Co., Ltd., Japan). The thermal stability of the dry samples were measured by thermogravimetric analysis (TGA, Model TA2100, TA Instruments, USA) under N2 at a heating rate of 10 °C/min from 50 to 650 °C. The drug loading and release rate were determined by UV–Vis spectrophotometry (UV–Vis, Agilent Co. Ltd., USA).
2.3 Synthesis of Fe3O4 nanoparticles
Oleic acid-modified Fe3O4 nanoparticles were synthetized by a classical coprecipitation method [23]. In a typical procedure of preparation, 2.4 g FeCl3·6H2O and 1.0 g FeCl2·4H2O were dissolved into 30 mL of deionized water, and then the mixture was heated to 80 °C with vigorous stirring for 30 min under the protection of N2. Later, 6 mL of ammonia hydroxide (25 wt%) was rapidly added and the color of mixture turned to black immediately, under the condition of continuous stirring for 30 min, 400 μL of oleic acid was added slowly. After that, the mixture was stirred for 3 h at 80 °C and then cooled naturally to room temperature. The black precipitated particles were collected by a magnet and washed with deionized water and ethanol several times, respectively. Fe3O4 nanoparticles were added in a vacuum drying oven for 24 h at 40 °C, then 58 mg dried Fe3O4 nanoparticles was dispersed in 7 mL of chloroform to form magnetic fluid and set aside.
2.4 Synthesis of magnetic mesoporous silica (Fe3O4@SiO2)
0.7 mL of as-prepared magnetic fluid was added into 10 mL of deionized water containing 0.15 g CTAB, the mixture was heated to 50 °C for 30 min under the condition of ultrasonic vibration to form emulsion system with the evaporation of chloroform. Then the obtained mixture was further heated to 70 °C for 10 min with stirring to evaporate residual chloroform, subsequently, 30 mL of deionized water, 10 mL of ethylene glycol and 1 mL of ammonia hydroxide (25 wt%) were added into the mixture. After 2 h of stirring at 70 °C, 500 μL of TEOS were added slowly and the mixture was continued to react for another 3 h. Finally, the product was collected by centrifugalization at 10,000 rpm and washed with deionized water and ethanol several times, respectively.
Magnetic silica was added into 30 mL of deionized water containing 60 mg ammonium nitrate at 60 °C for 1 h, and the above process was repeated 3 times to removal the templates. After that, the power was dried by vacuum drying at 50 °C for 12 h. Then, 100 mg dried power was added into 50 mL of isopropanol, after adding 70 μL of APTES, the mixture was heated to 80 °C for 12 h. Finally, Fe3O4@SiO2 was collected, washed and dried.
2.5 Drug loading
100 mg Fe3O4@SiO2 and 5 mg MTX was added into 30 mL of phosphate buffer solution (pH = 7.4) at 25 °C with string for 48 h. The drug-loaded nanoparticle (Fe3O4@SiO2-MTX) was collected by centrifugalization at 10,000 rpm and washed with deionized water and ethanol several times, respectively.
The embedding rate (%) E = (m1/m0) × 100 %, where m1 was the quality of MTX loaded in the carriers and m0 was the quality of MTX used in the reactions.
2.6 Preparation of Fe3O4@(SiO2-MTX)@AlOOH microspheres
Firstly, 5.65 g aluminum isopropoxide was dispersed into 50 mL of deionized water with stirring for 30 min at 85 °C, then HNO3 (1.0 M) was added into the mixture slowly until the pH value of the solution was kept in the range 3–4. Subsequently, the mixture was needed to continue the stirring process for another 2 h, and then slowly cooled to room temperature. After the evaporation of water, solid boehmite (AlOOH) was collected and the solid was ground into a powder, then 2.9 g boehmite was added into 50 mL of deionized water with stirring at 85 °C until the solid was completely dissolved, and 4.6 mL of HNO3 (1.0 M) was added into the solution slowly, the solution was continue heating at 85 °C with stirring for another 6 h. Finally, the solution obtained was naturally cooled to room temperature.
100 mg Fe3O4@SiO2-MTX was dispersed in 10 mL of AlOOH primer sol containing 5 mg MTX for 30 min with string, and followed by centrifugalization to separate Fe3O4@(SiO2-MTX)@AlOOH from the primer sol. Then the microspheres was washed with ethanol and dried in air for 30 min. The above process was repeated 5 times.
2.7 Preparation of Fe3O4@SiO2@LDH-MTX
0.15 g Mg(NO3)2·6H2O, 0.38 g NH4NO3 and 5 mg MTX were dissolved into 120 mL of deionized water. Then the as-prepared Fe3O4@(SiO2-MTX)@AlOOH was placed in above solution with stirring at 80 °C for 24 h. At last, the final nanoparticle (Fe3O4@SiO2@LDH-MTX) was collected by centrifugalization at 10,000 rpm and washed with deionized water and ethanol several times, respectively. In addition, the washed ethanol, deionized water and the initial reaction solution in this study all were collected, and the collected liquid was then detected via UV–Vis analysis at a wavelength of 290 nm.
2.8 In vitro release of MTX
Fe3O4@SiO2@LDH-MTX was equally divided into three portions (10 mg each) and dispersed into 5 mL of phosphate buffer solution (PBS) with different pH values of 7.4, 5.0 and 4.0, respectively. Then the mixtures were transferred to dialysis bags and the systems were soaked in 10 mL of the same buffer solution and placed in an oscillator at 37 °C. After oscillation for an appropriate time of interval (2, 4, 6, 8, 12, 24, 48, 72 h), 2 mL of soak solution outside the dialysis bag was taken out for UV–Vis analysis at a wavelength of 290 nm, while adding another 2 mL fresh PBS to maintain constance of soak solution volume. The drug release efficiency of Fe3O4@SiO2@LDH-MTX was measured by UV–Vis spectrophotometer, and the equation was described as follows: Drug release efficiency (%) R = (m2/m1) × 100 %, where m1 was the quality of MTX loaded in the carriers and m2 was the quality of MTX released from the carriers [7].
2.9 Cytotoxicity assay
Firstly, Hela cells were seeded into 24-well plates (2 × 104 cell/cm2). When the cell confluence reached around 60–70 %, the culture medium was replaced with 200 μL of fresh medium and 20 μL of free MTX, Fe3O4@SiO2@LDH or Fe3O4@SiO2@LDH-MTX at various concentrations in fresh cell culture medium was added, then Hela cells were further incubated at 37 °C for 24 h. Finally, 20 μL WST-1 solution was added to each well and the cells were incubated for another 2 h. For each well, enzyme linked immunosorbent assay (ELISA) instrument was used to measure the absorbance at a wavelength of 450 nm. Therefore, comparing the absorbance at 450 nm of the aforementioned wells and control wells containing only cell culture medium, the relative cell viability (%) was easily determined.
3 Results and discussion
The FT-IR spectra of Fe3O4@SiO2 and Fe3O4@SiO2@LDH were compared in Fig. 2. The absorption peaks at 582 cm−1 observed can be attributed to the Fe–O vibration of Fe3O4 nanoparticles [24] in the curves of Fe3O4@SiO2 and Fe3O4@SiO2@LDH. Furthermore, the absorption bands at 1032 and 778 cm−1 were also observed obviously, caused by the Si–O–Si vibration and Si–OH vibration, suggesting that silica was successfully coated on the surface of Fe3O4 nanoparticles. However, by contrast with Fe3O4@SiO2, the absorption peaks at 3438 and 1637 cm−1 in the curves of Fe3O4@SiO2@LDH were ascribed to the stretching and bending vibrations of O–H groups from the OH bonds and crystal water in the interlayer, indicating LDH was coated on the surface of silica. In addition, a new band at 1352 cm−1 was due to the presence of nitrate anions (NO3 −), caused by the precipitant of NH4NO3. Anions existed between the plate layers, and through ionic exchanging, anionic type drugs were exchange with NO3 −. Thus, a small amount of drugs were stored in the interlayer of LDH. Based on the above data analysis, it was primarily confirmed that Fe3O4@SiO2@LDH was successfully synthesized which can be used as drug carriers.

FT-IR spectra of Fe3O4@SiO2 and Fe3O4@SiO2@LDH
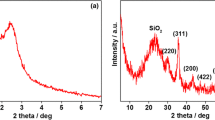
The X-ray diffraction (XRD) patterns of Fe3O4, Fe3O4@SiO2, Fe3O4@(SiO2-MTX)@AlOOH and Fe3O4@SiO2@LDH were recorded by X-ray diffractometer. As can be seen in Fig. 3, all samples exhibited typical diffractions of Fe3O4, including (220), (311), (400), (511) and (440) reflection planes, which was indicative of the presence of Fe3O4 crystallite. It was the reason for the carriers to express superparamagnetic behavior. After coating with the silica, a broad peak at 20°–30° was observed, indicating the amorphous nature of the silica coating. In addition, the XRD pattern of Fe3O4@(SiO2-MTX)@AlOOH was practically identical to that of Fe3O4@SiO2, suggesting MTX loaded in the channels of mesoporous silica and AlOOH coated on the surface of Fe3O4@SiO2 had a very small impact on the crystalline structure of Fe3O4. However, compared with Fe3O4@(SiO2-MTX)@AlOOH, (003), (006) and (110) reflection planes were clearly observed in the curves of Fe3O4@SiO2@LDH, It was typical diffractions of MgAl-LDH, reflecting the presence of LDH. In the course of the experiment, it was important to maintain the structure of Fe3O4, which would directly affect the targeting ability of the carriers.

XRD patterns of Fe3O4, Fe3O4@SiO2, Fe3O4@(SiO2-MTX)@AlOOH and Fe3O4@SiO2@LDH
The targeting ability of the carriers was derived from the magnetic responsivity, thus magnetic properties was recorded on vibrating sample magnetometer (VSM). As can be seen in Fig. 4, the saturation magnetization value of Fe3O4, Fe3O4@SiO2, Fe3O4@(SiO2-MTX)@AlOOH and Fe3O4@SiO2@ LDH-MTX were 75.19, 49.06, 34.26, 28.63 emu/g, respectively. The hysteresis loops confirmed the superparamagnetism of these nanoparticles, thus, drugs loaded in carriers would be transported to the designated location in the presence of an external magnetic field. In addition, a huge fall in the saturation magnetization values was observed among Fe3O4, Fe3O4@SiO2 and Fe3O4@(SiO2-MTX)@AlOOH, suggesting that the coating of silica and AlOOH had a great influence on the magnetic properties of the carriers. In contrary, AlOOH reacted with magnesium nitrate to form LDH had a relatively small influence. The results above showed that Fe3O4@SiO2@LDH-MTX possessed satisfied magnetic responsivity, which was important in terms of directional transportation and recycling of carriers.

VSM results of Fe3O4, Fe3O4@SiO2, Fe3O4@(SiO2-MTX)@AlOOH and Fe3O4@SiO2@LDH-MTX
Nitrogen sorption isotherms and the corresponding pore size distributions of Fe3O4@SiO2 were carried out to detect the porous structure. As can be shown in Fig. 5a, Fe3O4@SiO2 exhibited type IV adsorption–desorption isotherms with the H1 hysteresis loop, indicating that uniform mesopores were presented in Fe3O4@SiO2, which can be used to store anticancer drugs. In addition, according to the H1-type hysteresis loops, the pores of Fe3O4@SiO2 were cylindrical and open. However, the capillary condensation step of Fe3O4@SiO2 seemed to be not very steep, reflecting a not very evident ordered pore structure. Besides, starting from P/P0 = 0.9, a secondary condensation step was observed, due to the interparticle spaces or textual porosity formed after drying. Aside from that, the pore size distribution of Fe3O4@SiO2 determined by the traditional BJH method was shown in Fig. 5b, and the surface area, pore volume and pore diameter of Fe3O4@SiO2 were listed in Table 1, from which it can be seen that Fe3O4@SiO2 possessed large specific surface. Moreover, the pore size distribution plot showed that the average size of the pores was in the range of 1–4 nm, and the sharp peak centered at 2.84 nm. The surface area and pore volume of Fe3O4@SiO2 were measured to be 1095.898 and 1.915 cm3/g, respectively, revealing a huge potential applications in loading certain drugs or other biomolecules.

Nitrogen sorption isotherms (a) and corresponding pore size distributions (b) of Fe3O4@SiO2
The TEM images of Fe3O4 revealed that the average size of spherical nanoparticles was observed to be 10–15 nm (Fig. 6a), Fe3O4@SiO2, Fe3O4@(SiO2-MTX)@AlOOH and Fe3O4@SiO2@LDH were shown in Fig. 6b–d. From the TEM images, it can be seen that the average size of Fe3O4@SiO2 was observed to be 50–60 nm, the average size of Fe3O4@(SiO2-MTX)@AlOOH was 70–80 nm and the average size of Fe3O4@SiO2@ LDH was 90–100 nm, illustrating the coating thickness of AlOOH and LDH were about 20 and 30 nm, respectively. In addition, as can be seen in Fig. 6b, the obvious core–shell structure with cylindrical mesopores on the shell, which was a channel for drug loading, was observed and the shape of these nanoparticles was closed to spherical. Furthermore, AlOOH was coated on the surface of mesoporous silica and liked a film (Fig. 6c), while LDH was coated on the surface of mesoporous silica and appeared petaline shape (Fig. 6d). Thus, the TEM results visually illustrated that Fe3O4@SiO2@LDH were successfully synthesized, and further confirmed the analysis results of FT-IR and XRD.

TEM images of a Fe3O4, b Fe3O4@SiO2, c Fe3O4@(SiO2-MTX)@AlOOH and d Fe3O4@SiO2@LDH
The SEM images of Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2@ LDH were shown in Fig. 7. In Fig. 7a, it can be observed that the average particle size of Fe3O4 nanoparticles was about 10–15 nm in agreement with the results of the TEM analysis. Furthermore, in Fig. 7b, Fe3O4@SiO2 nanoparticles was observed to be spherical and the average particle size was 50–60 nm. After coating with LDH, the surface of Fe3O4@SiO2 appeared as jelly (Fig. 7c). More than that, the gelatinous microspheres were uniform both in size and shape, and LDH nanoplatelet grafted to the surface of SiO2 stacked together to form the edges of Fe3O4@SiO2@LDH. All of the above results were consistent with the results of TEM, and also proved that Fe3O4@SiO2@LDH was successfully synthesized to delivery drugs.

SEM images of a Fe3O4, b Fe3O4@SiO2 and c Fe3O4@SiO2@LDH
Figure 8 showed the energy dispersive spectrometer (EDS) images of Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2@ LDH. For all samples, Au (samples were treated by spray-gold), O and Fe elements were detected, caused by the presence of Fe3O4, which was the core of the composite. Furthermore, in Fig. 8b, c, Si elements were detected, due to the coating of silica. In contrast with Fe3O4@SiO2, Mg and Al elements were additional detected, illustrating the LDH layers were grafted on the surface of silica. The result above further proved that MgAl-LDH was coated on the surface of silica and it was identical with the XRD analysis.

The EDS images of a Fe3O4, b Fe3O4@SiO2 and c Fe3O4@SiO2@LDH
In order to confirm the thermal property of Fe3O4@SiO2, Fe3O4@(SiO2-MTX)@AlOOH,Fe3O4@SiO2@LDH and Fe3O4@SiO2@LDH-MTX, the TG analysis was adopted to estimate the amounts of MTX loaded in the carriers. As shown in Fig. 9, for all the samples, the weight loss in the range of 50–100 °C was caused by the loss of the physically adsorbed water and the structure water was lost in the range of 100–600 °C. In addition, contrast with Fe3O4@SiO2, Fe3O4@SiO2@LDH exhibited a weight loss of 25.13 %, corresponding to the loss of the LDH coated on the surface of Fe3O4@SiO2,and Fe3O4@(SiO2-MTX)@AlOOH exhibited a weight loss of 12.58 %, corresponding to the loss of AlOOH (coated on the surface of Fe3O4@SiO2) and MTX (loaded in the pipelines of mesoporous silica). Furthermore, compared with the weight loss of Fe3O4@(SiO2-MTX)@AlOOH, Fe3O4@SiO2@LDH-MTX showed additional 12.90 % weight loss, which was the change amount from AlOOH to LDH on the surface of carriers, and also it included the amount of MTX loaded in the interlayer of LDH, thus it explained that LDH was successful formed on the surface of carriers. In order to further determine whether the drug loaded in the carriers, and measure the loading amount of MTX for quantitative determination, UV–Vis analysis was used to verify the results of TGA.

TG analysis results of Fe3O4@SiO2, Fe3O4@(SiO2-MTX)@AlOOH, Fe3O4@SiO2@LDH and Fe3O4@SiO2MTX
Figure 10 showed UV–Vis spectra of MTX, Fe3O4@SiO2@LDH and Fe3O4@SiO2@LDH-MTX. As shown in Fig. 10, the characteristic UV–Vis absorption peaks of MTX were appeared at about 290 nm, and the similar peaks were observed in the curve of Fe3O4@SiO2@LDH-MTX, while there were no observed absorption peaks in the curve of Fe3O4@SiO2@LDH. Illustrating fully that MTX have been loaded in the carriers. Besides, MTX was inserted into nanotubes and interlayer, and the embedding rate of Fe3O4@SiO2 and LDH were 60.32 and 0.67 %, respectively. Furthermore 2.42 % of MTX was premature released from Fe3O4@SiO2 during the coating of LDH, all the results were basically consistent with the results of the TG analysis. Thus, the results above indicated that MTX was successfully loaded in the carriers, and Fe3O4@SiO2@LDH can be used as drug carriers to deliver MTX.

UV–Vis spectra of MTX, Fe3O4@SiO2@LDH and Fe3O4@SiO2@LDH-MTX
As can be seen in Fig. 11, a fast release happened within 8 h, and then followed by a relatively slow release rate until the end of assay, showing a significant sustained release effect. In addition, at pH 5.0 and 4.0, 55.27 and 66.16 % of MTX were released within 72 h, respectively, but at pH 7.4, the drug release rate was so low that only 20.91 % of MTX was released. Indicating that in vitro release of drugs from this system revealed pH sensitivity. Furthermore, the release rate of MTX loaded in the carriers increased with the decrease of pH value. Hence, the pH-sensitive LDH can be a great fracture to catch the purpose of drug sustained-release in tumor cells, while in normal cells to keep drugs and fail to release. So that, specific treatment was implemented and the drug delivery systems reduced the damage to human body effectively.

The release profiles of MTX from Fe3O4@SiO2-MTX
The cytotoxicity of free MTX, Fe3O4@SiO2@LDH and Fe3O4@SiO2@LDH-MTX were evaluated by WST-1 assay against Hela cancer cells. Figure 12 showed the related cytotoxicity situation, compared with MTX, no significant cytotoxicity can be observed for Fe3O4@SiO2@LDH, indicating that Fe3O4@SiO2@LDH can be regarded as safe drug delivery carriers. Then, compared with Fe3O4@SiO2@LDH, Fe3O4@SiO2@LDH-MTX exhibited higher toxicity and even higher than free MTX, it was because the magnetism of Fe3O4@SiO2 would help the internalization of Fe3O4@SiO2@LDH-MTX within cancer cell compartments, and the tendency of oxygen capture from the cell inspired Fe3+ to exhibit slightly bigger than Fe2+ [25]. All the results illustrated that Fe3O4@SiO2@LDH presented the great application potential for targeted drug delivery and Fe3O4@SiO2@LDH-MTX was available for magnetic targeted cancer therapy.

Cytotoxicity evaluation of MTX solution, suspensions of Fe3O4@SiO2@LDH and of Fe3O4@SiO2@LDH-MTX by WST-1 assay against Hela cells after 24 h treatment
4 Conclusion
To sum up, Fe3O4@SiO2@LDH nanocomposites with bioaffinity and targeting performance were successfully prepared for the delivery of MTX in this study. All the results demonstrated that these Fe3O4@SiO2@LDH nanocomposites have the suitable size around 100 nm, superparamagnetic behavior and reasonable loading rate. Besides, the prepared Fe3O4@SiO2@LDH-MTX systems exerted high pH-sensitive for drug delivery in vitro and the release rate of the drug delivery systems was 66.16 % within 72 h at pH 4.0. Furthermore, under the double action of mesoporous silica and LDH, 60.32 % of MTX was loaded in the mesoporous silica and 0.67 % of MTX was loaded in LDH. Therefore, we have enough reasons to believe that Fe3O4@SiO2@LDH-MTX would present a great potential application in biological target cancer therapy.
References
M.C. Daniel, D. Astruc, Chem. Rev. 104, 293–346 (2004)
E. Ruoslahti, S.N. Bhatia, M.J. Sailor, J. Cell. 188, 759–768 (2010)
J. Wu, W. Jiang, S.S. Xu, Y.J. Wang, R. Tian, Sens. Actuators B: Chem. 211, 33–41 (2015)
I.I. Slowing, J. Mater. Chem. 20, 7924–7937 (2010)
S.S. Huang, C.X. Li, Z.Y. Cheng, Y. Fan, P.P. Yang, C.M. Zhang, K.Y. Yang, J. Lin, J. Colloid Interface Sci. 376, 312–321 (2012)
X. Tian, Z. Dong, R. Wang, J. Ma, Sens. Actuators, B 183, 446–453 (2013)
J. Wu, Y.J. Wang, W. Jiang, S.S. Xu, R.B. Tian, Appl. Surf. Sci. 321, 43–49 (2014)
K.H. Shim, J. Hulme, E.H. Maeng, M.K. Kim, S.S. An, Int. J. Nanomed. 9, 207–215 (2014)
J. Wang, R.R. Zhu, B. Gao, B. Wu, K. Li, X.Y. Sun, H. Liu, S.L. Wang, Biomaterials 35, 466–478 (2014)
Y. Deng, D. Qi, C. Deng, X. Zhang, D. Zhao, JACS 130, 28–29 (2008)
Q. Liu, J.X. Zhang, W. Sun, Q.R.B. Xie, W.L. Xia, H.C. Gu, Int. J. Nanomed. 7, 999–1013 (2012)
X. Guo, F. Mao, W. Wang, Y. Yang, Z. Bai, A.C.S. Appl, Mat. Interfaces 7, 14983–14991 (2015)
J.L. Vivero-Escoto, I.I. Slowing, B.G. Trewyn, V.S.-Y. Lin, Small 6, 1952–1967 (2010)
A.K. Gupta, M. Gupta, Biomaterials 26, 3995–4021 (2005)
C.E. Myers, W.P. Mcguire, R.H. Liss, I. Ifrim, K. Grotzinger, R.C. Young, Science 197, 165–167 (1977)
L.E. Gerweck, K. Seetharaman, Cancer Res. 56, 1194–1198 (1996)
J. Zhang, X. Li, J.M. Rosenholm, H.C. Gu, J. Colloid Interface Sci. 361, 16–24 (2011)
Y. Chen, H.G. Chen, S.J. Zhang, F. Chen, L.X. Zhang, J.M. Zhang, M. Zhu, H.X. Wu, L.M. Guo, J.W. Feng, J.L. Shi, Adv. Funct. Mater. 21, 270–278 (2011)
C.Y. Lai, B.G. Trewyn, D.M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija, V.S. Lin, J. Am. Chem. Soc. 125, 4451–4459 (2003)
K.M. Tyner, S.R. Schiffman, E.P. Giannelis, J. Control Release 95, 501–514 (2004)
A. Tarnawski, R. Pai, R. Itani, F.A. Wyle, Digestion 60, 449–455 (1999)
Z. Gu, A.C. Thomas, Z.P. Xu, J.H. Campbell, G.Q. Lu, Chem. Mater. 20, 3715–3722 (2008)
J.H. Wang, S.R. Zheng, Y. Shao, J.L. Liu, Z.Y. Xu, J. Colloid Interface Sci. 349, 293–299 (2010)
J. Wu, S.S. Xu, W. Jiang, Y. Shen, M. Pu, Biotechnol. Lett. 3, 585–591 (2015)
J. Varshosaz, H. Sadeghi-aliabadi, S. Ghasemi, B. Behdadfar, Biomed. Res. Int. 2013, 1–16 (2013)
Acknowledgments
The authors are grateful for National Science Foundation of China (No. 50972060), the Fundamental Research Funds for the Central Universities (No. 30920130112003) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jiang, W., Wu, J., Tian, R. et al. Synthesis and characterization of magnetic mesoporous core–shell nanocomposites for targeted drug delivery applications. J Porous Mater 24, 257–265 (2017). https://doi.org/10.1007/s10934-016-0259-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-016-0259-z