
Abstract
The inflammatory cytokine, interleukin-2 (IL-2), is an important regulator of cellular functions. This relatively less studied member of the interleukin protein family is responsible for multiple immuno-modulatory and immuno-stimulatory tasks, like T cell activation, triggering of natural killer cells, inflammation, as well as proliferation and progression of autoimmune diseases and cancers. In this communication we report the temporally variant structural aspects of the IL-2 ligand and its receptor interfaces, based on the available crystal structures. The intended goal of this effort is to generate simulated results that could potentially aid the designs of novel structure based therapeutics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The members of the interleukin (IL) family, consisting of IL-1 to IL-41, form a structurally and functionally diverse group of cytokines. Among these, interleukin-2 (IL-2), an inflammatory cytokine, is an essential regulator for cellular functioning [1,2,3,4]. The IL-2 protein is responsible for multiple immunomodulatory tasks, such as the activation of T cells, triggering of natural killer cells, inflammation, and proliferation/pathogenesis of serious illnesses like autoimmune diseases and cancers [5,6,7,8,9]. The IL-2 ligand-receptor (IL-2/IL-2R), which is the central system of our present investigation, dictates various immuno-regulatory/stimulatory reactions involving complex cellular signaling processes. The IL-2 binds to the heterotrimeric IL-2R to generate the signaling complex that initiates the signal-transduction process [10,11,12].
We have previously examined the structures and structure–function relationships of several physiologically significant proteins linked to cellular signaling [13,14,15,16,17,18]. Using exploratory molecular dynamics (MD) simulation [19, 20], the present work aims at probing the molecular structure and the time-dependent developments of IL-2 and its receptors. This study is motivated by the observation that, these types of structural analyses of ligand/receptor proteins may potentially assist the development of new therapeutic designs [21, 22]. As part of the investigation, we also examine the interfacial protein–protein interactions (PPIs) of the IL-2 ligands and their receptors, along with the structural and conformational stabilities of the wild type (wt) protein and protein complexes. The associated signaling pathways and their biological implications are explored as well.
2 Experimental Section
2.1 Model Selection for Molecular Simulation
For the purpose of simulation, we use 3INK.PDB (chain C), based on the X-ray crystallographic structure of wt IL-2, developed by D. B. McKay [5]. For comparison of results, we choose as a reference system, the IL-2Rα-receptor bound IL-2, 1Z92.PDB, reported by Rickert et al. [6]. We also simulate the tetrameric unit of 2B5I.PDB system, for which, the IL-2 (chain A) bound IL-2α (chain D), β (chain B), and common γ (chain C) receptor subunits have been described by Wang et al. [7]. As an extension of the main investigation, in the Electronic Supplementary Material (ESM), we briefly examine the 2ERJ.PDB system reported by Stauber et al. [8]. 2ERJ has two trimeric units of Il-2 bound IL-2 receptors (α, β and γ), and represents a complex structure composed of IL-2 (chains D and H) bound hetero-trimeric IL-2Rα (A and E), β (Β and F), and common γ chains (C and G).
2.2 Software for Simulation Data Analysis
NAMD and VMD software packages are used for molecular dynamics simulation and simulation trajectory analyses [23, 24]. The Cologne University Protein Stability Analysis Tool (CUPSAT) is used to analyze IL-2’s stability upon mutation [25], and Cytoscape is employed to analyze protein–protein interaction network for IL-2 signaling cascades [26]. 3D graphics are developed with Discovery Studio Visualizer [27], while Origin is used to process the data and to generate the plots.
2.3 Experimental MD Simulation Setup
Initially the protein structure files were created using autoPSF graphical user interface of VMD. Energy minimization was performed using NPT ensemble for 10,000 steps. Before energy minimization, solvated and neutralized IL-2, IL-2/IL-2R structures were obtained using the solvate and auto-ionize modules of VMD. The production run was performed for 20 ns at 298 K using the NVT ensemble with the periodic boundary condition. The particle mesh ewald (PME) was set as active. Additional experimental details of the present work are described elsewhere [14, 15].
3 Results
Figure 1 shows a schematic representation of the IL-2/IL-2R signaling cascades, generated using the cytoscape protein–protein interaction software [26]; the interaction networks are imported form the reactome database [28]. The IL-2 ligand and receptor subunits are highlighted in Fig. 1a, where the nodes identify the individual proteins, and the edges represent their associations. The workflow interpretation of the IL-2/IL-2R network architecture, as obtained from the proteins’ UniProt accession ids, is presented in Fig. 1b. The main UniProt accession id for Il-2, P60568 is indicated in large fonts. Figure 2 displays the major signaling cascades triggered by the IL-2/IL-2R system. The impact of IL-2 as a signaling molecule and its role in various disease states will be discussed later, in Sects. 4.1 and 4.4 of this report.

(Color online only) a Schematics of the IL-2/IL-2R signaling pathways. b The workflow view of the IL-2/IL-2R network, based on the protein’s UniProt Accession ID

(Color online only) Drawing illustrating the major signaling cascades of IL-2/IL-2R
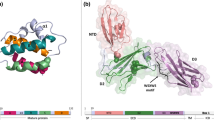
Figure 3a presents the secondary structure of wt IL-2. 3INK, the X-ray crystallographic structure of wt apo IL-2 [5], is a homo dimer where each subunit contains 133 amino acid (aa) residues. Each subunit of 3INK contains four antiparallel α helices joined by looped regions. Three cell surface receptors (IL-2Rα, -β and -γ) simultaneously bind to the IL-2 ligand protein with varied affinities [29]. The 1L-2/IL-2Rαβγ trimer-complex initiates the signal transduction processes. Mutation induced free energy changes (ΔΔG) for the wt IL-2 protein 3INK.PDB were determined by using the CUPSAT server [25]; these results are tabulated in Fig. 3b and c. Contributions of both chemical and thermal denaturation processes are included in these results. In most cases, these mutations are found to have an overall stabilizing effect where the ΔΔG value, as calculated in the simulation, is positive. The torsion angles observed in these cases are favorable for single amino acid (aa) mutation.

(Color online only) Ribbon diagram of wt IL-2 protein. a Secondary structure of 3INK.PDB (red) is displayed. b and c Mutation induced stability changes in wt IL-2 protein 3INK.PDB. We have predicted the ΔΔG changes using. b Chemical denaturation and c thermal denaturation methods
As reported by by Rickert et al. [6], 1Z92.PDB, the X ray crystallographic structure of IL-2 bound IL-2Rα, is a heterodimer. Its chain A (mostly α helical) is composed of a 133 residue long IL-2, and chain B represents a 219 aa long IL-2Rα. The secondary structure of ligand bound IL-2R, as shown in Fig. 4a, suggests that, the IL-2 ligand primarily interacts with its receptor’s hydrophobic surface. Further details of these interfacial residues and their interactions are described later in Sect. 4.2.

(Color online only) Ribbon diagram of IL-2 protein, and IL-2 bound receptors. a Structure of 1Z92.PDB where the ligand-receptor system of IL-2 (yellow) and IL-2R (green) is shown. b Structure of 2B5I.PDB, including the IL-2 ligand bound (Chain A; pink) α (Chain D; violet), β (Chain B; blue), and γ (Chain C; cyan) receptor subunits. c Structure of one tetrameric unit of 2ERJ.PDB, where the IL-2 ligand (Chain D, brown) bound hetero-trimeric IL-2Rα (Chain A, dark green), β (Chain Β, light green), and common γ (Chain C, yellowish green) subunits are displayed
2B5I.PDB (Fig. 4b) is a hetero-tetrameric complex, and as indicated in its X-ray crystallographic structure reported by Wang et al. [7], this protein contains the IL-2 ligand (IL-2L; chain A) bound IL-2Rα (chain D) along with the β (chain B) and common γ (chain C) subunits. In this case, the lengths, measured in aa units, for the ligand (A), the IL-2Rα (D), -β (B), and the common -γ (C) chains are 133, 217, 214 and 199, respectively. The PDB structure also shows that, the IL-2Rβ and -γ subunits are separated by a distinct groove where the specific IL-2 ligand bound site is located. There are four inter-subunit interaction sites for this system, as described by Wang et al. These are observed between the pairs, IL-2 ligand (IL-2, chain A)/IL-2Rα (chain D), IL-2/IL2Rβ (chain B), IL-2/IL-2Rγ (chain C) and also between IL-2Rβ (chain B)/IL-2Rγ (chain C). However, no interactions are detected between IL-2Rα and IL-2Rβ or -γ.
In this context, we also examine 2ERJ.PDB, which is another complex of IL-2 ligand (Chains D and H) bound hetero-trimeric unit composed of the IL-2Rα (Chains A and E), β (Β and F), and common γ (Chains C and G) subunits [8]. The crystallographic unit of the monomeric-ligand bound trimeric receptor structure of 2ERJ is displayed in Fig. 4c.
Figure S1 of ESM and Table 1 summarize the interfacial residues of the IL-2 ligands and receptors that are likely participants of the binding actions in 1Z92.PDB. The interfacial interactions in 1Z92.PDB are tabulated in ESM Table S1 while ESM Fig. S2 schematically shows the operative interactions between the relevant interface residues. The interfacial residues of the IL-2 ligand and the receptors of 2B51.PDB are depicted in Fig. S3 and tabulated in Table 2. As reported by Wang et al. [7], the interacting ligand-receptor interfaces in 2B51 participate in several van der Waals type interactions. Other non-bond interactions between the interfacial residues are tabulated in ESM Table S2. The water mediated interactions in 2B5I have already been described by Wang et al., and hence are not included in Table S2.
The largely unwavering temporal profile of the data in Fig. 5a indicates that, all the protein and protein complexes examined in these MD simulations are fairly stable with time. The wt IL-2 protein exhibits the lowest variation in this group, and the ligand bound receptor structures are also practically stable. 1Z92, one of the systems in Fig. 5a does not adequately reach convergence. To address this observation, we note that, extended run times (1–5 μs) are often necessary to observe complete convergences of biomolecular systems [30], although in some cases, a plateau cannot be reached even after rather prolonged simulations of ~ 1 ms [31, 32]. Convergence/stability criteria for such systems can often be phenomenologically characterized in terms of RMSD values. If the occurrence of larger RMSD values is limited to a relatively short time span, or if the RMSD difference measured between repeated simulations is moderate, the system in general can be considered convergent/stable.

(Color online only) a Typical all-atom RMSD plots for the wt IL-2 and ligand–bound IL-2R protein systems. b RMSD plots of the ligand-receptor interfacial residues in IL-2 bound receptor systems. c Alpha carbon RMSF plots for the wt IL-2 and ligand–bound IL-2R system. d Alpha carbon RMSF plots for individual subunits within ligand-bound trimeric IL-2R system
In view of the above considerations, and for a quick check of data repeatability for the apparently non-converging system, we performed another 15 ns exploratory simulation using 1Z92.PDB. The supplementary Fig S4 displays the RMSD data overlaying the two sets of repeated results on a common time-axis of 15 ns. The calculated maximum RMSD values for these two sets are 4.46 Å (first run) and 3.96 Å (repeated run). Thus the RMSD variations between the two cases are relatively small (within < 12% of each other’s values), and can be attributed to simulation artifacts. The observed agreement between the RMSD values for the two consecutive simulations demonstrates repeatability of results. Furthermore, the standard deviations (SDs) of the two sets of RMSD values are in relatively close agreement with each other, with values of 0.356 Å and 0.311 Å, for the first and second trials, respectively. These < 1 Å SD values are indicative of mutually agreeing results [33].
Figure 5b presents the RMSD values, calculated for the interfaces within the ligand-receptor systems. These data indicate that, the interfacial residues in the ligand–receptor system are overall stable as functions of time and, that the ligand protein binds to the receptor protein under mostly stable conditions. Figure 5c shows the root mean square fluctuations (RMSF) of the ligand proteins and the receptor proteins within the ligand–receptor systems. These time averaged fluctuations, taken as functions of residue numbers, indicate that, the wt protein is most stable among the systems compared here. In 1Z92.PDB, the ligand chain shows higher RMSF values than those of the receptor chain, and also exhibits some variations at both the bounding edges of the protein. These higher RMSFs observed at the terminal points can be associated with the elevated (time-specific, aa averaged) RMSD values of the protein complexes. Figure 5d shows the simulated RMSF data obtained within the 2B5I chains. In this case, subunits A and D are, respectively, the most stable and least stable systems among those compared.
The secondary structure of 3INK is displayed in Fig. 6. 3INK is fairly stable and no major secondary structural variations are observed although, some minor variations (α-helix to 3–10 helices) are detected near the residue 55. Figure 7 indicates a series of conformational changes within the interfacial residues of ligand-receptor complexes. The overall system stability within the 2B5I adjacent-interfaces appears to be relatively prevalent. These observations are consistent with those made in Fig. 4b, where the RMSD values of the 2B5I interfacial residues were more stable than the 1Z92 interfacial areas. In general the secondary structures of the IL-2/IL-2R interfaces in both systems remain stable over the simulation time.

(Color online only) a Time-series of proteins’ secondary structure-changes in wt IL-2 ligand protein, 3INK.PDB. b The color-code explanation of protein’s secondary structures

(Color online only) Time-series of proteins’ secondary structure-changes at selected interfacial areas of Il-2/IL-2R. a Secondary structure-changes of selected Chain A residues at the interface of 1Z92.PDB. b Secondary structure-changes of selected Chain B residues at the interface of 1Z92.PDB. c Secondary structure-changes of selected Chain A residues at the interface of 2B5I.PDB. d Secondary structure-changes of selected Chain B residues at the interface of 2B5I.PDB. e Secondary structure-changes of selected Chain C residues at the interface of 2B5I.PDB. f Secondary structure-changes of selected Chain D residues at the interface of 2B5I.PDB. These interfacial residues are described in the Tables 1 and 2. The color-code explanation of proteins’ secondary structures are displayed in Fig. 6b
The overall quality of the molecular models can be further assessed in terms of the choices of atomic coordinates used in the models. The resolution and the R-value (residual factor) are two key factors in this regard for X ray crystallographic structural models. The starting structures for 3INK, 1Z92 and 2B5I have the resolutions of 2.5 Å, 2.8 Å and 2.3 Å respectively. The corresponding R-values (Rfree/Rwork) for the three structures are NA/0.202, 0.280/0.233 and 0.269/0.223. Thus Rwork ≤ (Resolution/10) in all cases, and Rfree also mostly meets this criterion. These results are indicative of the overall efficacy of the three model systems.
4 Discussion
4.1 Biological Implication of IL-2 Systems
IL-2, a predominantly α helical protein, usually exists in a monomeric form. It is an immuno-modulatory cytokine with a critical role in the regulation of innate and adaptive immune responses. The hetero-trimeric IL-2R is composed of three subunits: IL-2 specific subunit α (CD25), subunit β (CD122), and a common subunit, γ (CD132), located on the cell-surface area. The IL-2Rβ and -γ subunits are common to the two closely resembled receptors, IL-2R and IL-15R [34]. The γ subunit of IL-2R is usually shared among some other IL receptor family members including IL-4, -7, -9, -15 and -21, and hence is referred to as common cytokine γ (γc) [11, 12].
The receptor bound IL-2 protein complexes were introduced by Garcia and coworkers [6, 7]. While the binding of IL-2 to IL-2Rα initiates the low affinity ligand-receptor complex formation, the ligand bound trimeric IL-2Rαβγ induces the ultimate signaling cascade. The heterotrimeric receptor bound form of IL-2 ligand initiates the Janus kinase 1/3 (JAK1/3) group of signaling pathways; these include the JAK/signal transducer and activator of transcription (JAK/STAT) and the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK); both are responsible for cell growth/proliferation and survival. The downstream effects of phosphatidylinositol 3kinase/protein kinase B (PI3K/PKB or Akt) signaling pathway play a key role in the regulatory process of a normal cell cycle.
4.2 Protein–Protein Interfaces and Interactions Between Crucial Residues
The inhibitors of PPIs are important for drug designing processes, and computational techniques help to identify the potent inhibitors of these types [35,36,37]. For instance, engineered monoclonal antibodies (mAbs) play a vital role in disrupting key IL-2 regulatory cascades and thus, the mAb function is relevant for immunotherapeutic drug developments. PPIs are governed by hydrophobic effects, by the nature of the associated hydrogen bonds, and also by various electrostatic, ionic and covalent interactions between the proteins’ interfaces [38,39,40]. The PPIs observed between ligand–receptor subunits of 1Z92.PDB and 2B5I.PDB include hydrophobic interactions, as well as interactions of hydrogen bonds (conventional and/or carbon-hydrogen bonds), salt-bridges, pi-alkyl, pi-cation, alkyl, and donor–donor/acceptor–acceptor pairs.
In the following, we discuss the specific interfacial features responsible for the protein–protein complexes in the IL-2/IL-2R systems. The Phe42, located on the loop region of the ligand surface of 1Z92.PDB is a key residue for receptor binding, as this residue strongly interacts with the receptor-residues through van der Waals and pi-alkyl interactions. Likewise, Leu72 is another IL-2 residue that exhibits strong alkyl and van der Waals interactions with the receptor chain. Met25, Leu42 and Tyr43 are the other residues of IL-2R, linked to the ligand binding details of IL-2 [6]. These latter residues are characterized by a range of inter-chain H-bonds (Leu42), alkyl/pi-alkyl (Met25, Leu42), acceptor–acceptor (Tyr 43) and van der Waals type interactions. The strong inter-subunit interactions within the 2B51 (described in ESM Table S2), make this protein more stable than 1Z92. Additionally, the cooperative subunit arrangements within the 2B5I [7] appear to play a role in making its time dependent structure somewhat steadier than the structure of 1Z92.
4.3 Molecular Dynamics Simulations
MD simulations were performed to assess the stabilities of the relevant proteins in their individual and coupled configurations [41,42,43]. In addition to simulating the wt protein, 3INK.PDB and the hetero-dimeric ligand-receptor system, 1Z92.PDB, we have carried out a simulation for the ligand bound receptor trimer, 2B5I.PDB. These analyses have been based on the representative crystal structures in combination with previously established modeling approaches of molecular dynamics [44]. In this setting, we have also highlighted the implications of these modeled molecular structures in determining the overall time dependent stabilities of wt IL-2 and IL-2/IL-2R.
Based on the results in Fig. 5a, we can postulate that the ligand bound receptor trimer is more stable/steady compared to the ligand bound receptor monomer. The inter-subunit interactions likely support the relatively low RMSD values observed for the ligand bound receptor trimer 2B5I. While Fig. 5b shows somewhat stable interfaces, the 1Z92 interface is comparatively less settled than 2B5I. The presence of interface loops in 1Z92 could be related to the presence of slight instability. PPIs are the likely factors responsible for making the tetrameric 2B51 interface more stable than the dimeric interface of 1Z92.
Based on the RMSD results, the interface of the D chain within 2B51 appears somewhat less stable. The smaller subunit interfacial area of chain D constrains this chain’s contact regions; this configurational restriction of subunit D can be associated with its lower stability. The RMSF values of chain A within 2B51 is more non-transitory compared to the other three chains. The twisted α-helical compact A subunit is surrounded by three other subunits. The interactions with the other three chains contribute to making the subunit A most stable.
The RMSF values of the B and C subunits exhibit essentially similar stabilities. These two subunits interact with each other and also with subunit A. In general the irregular “turn and loop” regions of proteins account for the greater flexibility, but tend to be relatively less stable. Based on Figs. 5, 6, and 7 we can infer that the overall structure of the wt protein is basically stable and conserved during the simulation time. The secondary structure of ligand–receptor interface in 2B5I is most stable due to extensible inter subunit interactions. Nevertheless the low-resolution and lower Rfree value for 2B5I could be associated with the latter’s relatively more specified structure.
The above results (in combination with those presented in the ESM) provide a computer simulated overall framework to identify the ligand–receptor interfaces of the protein complexes, and also help to assess their stabilities with time. Further extensions of such simulations could lead to, for example, a comprehensive approach to cataloging dynamically stable PPI regions. This in turn could be utilized for designing more stable protein variants, or targeted therapeutic agents tailored to various interfacial receptor stabilities.
4.4 Clinical Implications of IL-2/IL-2R Signaling in Disease Propagation and Design of Targeted IL-2 or IL-2R Therapeutics for Various Treatments
In the following we briefly note the role of IL-2 in immuno-modulatory or immuno-inhibitory therapies [45,46,47]. Since IL-2 serves to prompt autoimmune diseases and chronic inflammations, there are now growing interests among pharmaceutical industries to develop formulations for potent IL-2 and IL-2R mutants, or for IL-2/mAb complexes with greater effectiveness [48]. A Phe to Ala mutant variant NMR structure of IL-2 at position 42 has been developed by Mott et al. [49], and has been described by other authors as a mutant variant that would selectively bind to IL-2Rβ and not to IL-2Rα [50]. Likewise, Levin et al. has reported an IL-2 ‘superkine’ that binds to IL-2Rβ with preferentially greater affinity [51].
Several IL-2 and IL-2R based therapies are in their clinical phases at this time, among which, anti-IL-2 daclizumab® (Dac, Biogen.Inc and AbbVie) has been used until recently. Dac, is a human mAb that interacts with IL-2Rα and thus interrupts the coordinated JAK/STAT pathway. Basiliximab® (Novartis Inc.) is another human/murine chimeric mAb that selectively binds to IL-2Rα and blocks the binding between IL-2 and IL-2Rα. Aldesleukin® (Novartis Inc.), is a recombinant form of human IL-2 (rIL-2) used in renal cell cancer and melanoma treatments. Recent studies have further demonstrated that, selective bindings between IL-2 variants and their receptor subunits act to interrupt the essential steps in IL-2 signaling cascades [52, 53]. Interruption of the IL-2/JAK and JAK/STAT cascades by using suitable selective IL-2 and JAK inhibitors have been linked to widespread implications in next generation drug development [54].
5 Conclusions
Appropriate identification of protein-drug-targets is essential for new drug development. The structure based approach to probing the inter-subunit/interfacial stabilities of proteins can be particularly useful in this regard, since these analyses may help to categorize various cellular functions and corresponding innovative treatments. In this way, therapeutic approaches based on computational models can supplement lab-based investigations and clinical studies in a cost-effective way to facilitate drug designs for promoting cellular functions. However, this computational approach to diagnosing diseases and tailoring their treatments is still a relatively underexplored branch of the medical field. Specifically, the modeling approach that forms the basis of the present study can potentially play a significant role in understanding various pathological and physiological conditions and thus, can facilitate the development of future healthcare strategies and inventions.
Abbreviations
- aa:
-
Amino acid
- IL:
-
Interleukin
- IL-2:
-
Interleukin-2
- IL-2R:
-
Interleukin-2 receptor
- MD:
-
Molecular dynamics
- PDB:
-
Protein Data Bank
- RMSD:
-
Root mean square deviation
- RMSF:
-
Root mean square fluctuation
- TH :
-
T helper
References
Smith KA (1988) Interleukin-2: inception, impact, and implications. Science 240:1169–1176
Paul WE, Seder RA (1994) Lymphocyte responses and cytokines. Cell 76:241–251
Rickert M, Boulanger MJ, Goriatcheva N, Garcia KC (2004) Compensatory energetic mechanisms mediating the assembly of signaling complexes between interleukin-2 and its alpha, beta, and gamma(c) receptors. J Mol Biol 339:1115–1128
Spolski R, Li P, Leonard WJ (2018) Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol 18:648–659
McKay DB (1992) Response. Science 257:412–413
Rickert M, Wang X, Boulanger MJ, Goriatcheva N, Garcia KC (2005) The structure of interleukin-2 complexed with its alpha receptor. Science 308:1477–1480
Wang X, Rickert M, Garcia KC (2005) Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors. Science 310:1159–1163
Stauber DJ, Debler EW, Horton PA, Smith KA, Wilson IA (2006) Crystal structure of the IL-2 signaling complex: paradigm for a heterotrimeric cytokine receptor. Proc Natl Acad Sci USA 103:2788–2793
Liao W, Lin JX, Leonard WJ (2011) IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol 23:598–604
Kishimoto T, Taga T, Akira S (1994) Cytokine signal transduction. Cell 76:253–262
Rochman Y, Spolski R, Leonard WJ (2009) New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol 9:480–490
Wang X, Lupardus P, Laporte SL, Garcia KC (2009) Structural biology of shared cytokine receptors. Annu Rev Immunol 27:29–60
Roy U (2016) Structural biology of tumor necrosis factor demonstrated for undergraduate instruction by computer simulation. Biochem Mol Biol Educ 44:246–255
Roy U (2017) Structural modeling of tumor necrosis factor: a protein of immunological importance. Biotechnol Appl Biochem 64:454–463
Roy U (2019) 3D modeling of tumor necrosis factor receptor and tumor necrosis factor-bound receptor systems. Mol Inf 38:1–13. https://doi.org/10.1002/minf.201800011
Roy U, Luck LA (2007) Molecular modeling of estrogen receptor using molecular operating environment. Biochem Mol Biol Educ 35:238–243
Roy U, Luck LA (2011) Cysteine residues in heavy metal binding proteins: structural insights and comparison with leucine binding protein. JCCE 5:238–243
Roy U, Woods AG, Sokolowska I, Darie CC (2014) Utility of computational structural biology in mass spectrometry. In: Woods AG, Darie CC (eds) Advancements of mass spectrometry in biomedical Research. Springer, Cham, pp 107–128
Hedayati MH, Norouzian D, Aminian M, Teimourian S, Ahangari Cohan R, Sardari S, Khorramizadeh MR (2017) Molecular design, expression and evaluation of PASylated human recombinant erythropoietin with enhanced functional properties. Protein J 36:36–48
Srinivasan E, Rajasekaran R (2019) Computational investigation on electrostatic loop mutants instigating destabilization and aggregation on human SOD1 protein causing amyotrophic lateral sclerosis. Protein J 38:37–49
Bosco DA, LaVoie MJ, Petsko GA, Ringe D (2011) Proteostasis and movement disorders: Parkinson’s disease and amyotrophic lateral sclerosis. Cold Spring Harb Perspect Biol 3:a007500
Jiang Z, Vasil AI, Vasil ML, Hodges RS (2014) “Specificity determinants” improve therapeutic indices of two antimicrobial peptides Piscidin 1 and Dermaseptin S4 against the Gram-negative pathogens Acinetobacter baumannii and Pseudomonas aeruginosa. Pharmaceuticals (Basel, Switzerland) 7:366–391
Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
Humphrey W, Dalke A, Schulten K (1996) VMD. J Mol Graph 14:33–38
Parthiban V, Gromiha MM, Schomburg D (2006) CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res 34:W239–W242
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Discovery Studio Visualizer (2012) Discovery studio modeling environment, release 3.5. Accelrys Software Inc., San Diego Release 4.5, Dassault Systèmes BIOVIA, San Diego: Dassault Systèmes, 2015
Fabregat A, Korninger F, Viteri G, Sidiropoulos K, Marin-Garcia P, Ping P, Wu G, Stein L, D’Eustachio P, Hermjakob H (2018) Reactome graph database: efficient access to complex pathway data. PLoS Comput Biol 14:e1005968
Wilson CG, Arkin MR (2011) Small-molecule inhibitors of IL-2/IL-2R: lessons learned and applied. Curr Top Microbiol Immunol 348:25–59
Galindo-Murillo R, Roe DR, Cheatham TE 3rd (2015) Convergence and reproducibility in molecular dynamics simulations of the DNA duplex d(GCACGAACGAACGAACGC). Biochem Biophys Acta 1850:1041–1058
Zuckerman DM (2011) Equilibrium sampling in biomolecular simulations. Annu Rev Biophys 40:41–62
Genheden S, Ryde U (2012) Will molecular dynamics simulations of proteins ever reach equilibrium? Phys Chem Chem Phys 14:8662–8677
Zhao Y, Zeng C, Massiah MA (2015) Molecular dynamics simulation reveals insights into the mechanism of unfolding by the A130T/V mutations within the MID1 zinc-binding Bbox1 domain. PLoS ONE 10:e0124377
Bachmann MF, Oxenius A (2007) Interleukin 2: from immunostimulation to immunoregulation and back again. EMBO Rep 8:1142–1148
Sheinerman FB, Giraud E, Laoui A (2005) High affinity targets of protein kinase inhibitors have similar residues at the positions energetically important for binding. J Mol Biol 352:1134–1156
Zaidman D, Wolfson HJ (2017) Protein-peptide interaction design: PepCrawler and PinaColada. Methods Mol Biol 1561:279–290
Redwan EM, AlJaddawi AA, Uversky VN (2019) Structural disorder in the proteome and interactome of Alkhurma virus (ALKV). Cell Mol Life Sci 76:577–608
Berliner LJ (1984) Structure-function relationships in human alpha- and gamma-thrombins. Mol Cell Biochem 61:159–172
Salopek-Sondi B, Skeels MC, Swartz D, Luck LA (2003) Insight into the stability of the hydrophobic binding proteins of Escherichia coli: assessing the proteins for use as biosensors. Proteins 53:273–281
Gray NW, Zhorov BS, Moczydlowski EG (2013) Interaction of local anesthetics with the K (+) channel pore domain: KcsA as a model for drug-dependent tetramer stability. Channels (Austin, Tex) 7:182–193
Roy U (2016) Structural characterizations of the Fas receptor and the Fas-associated protein with death domain interactions. Protein J 35:51–60
Roy U, Woods AG, Sokolowska I, Darie CC (2013) Structural evaluation and analyses of tumor differentiation factor. Protein J 32:512–518
Ibrahim MAA, Hassan AMA (2018) Comparative modeling and evaluation of leukotriene B4 receptors for selective drug discovery towards the treatment of inflammatory diseases. Protein J 37:518–530
Bamborough P, Hedgecock CJ, Richards WG (1994) The interleukin-2 and interleukin-4 receptors studied by molecular modelling. Structure (London, Engl: 1993) 2:839–851
Arenas-Ramirez N, Woytschak J, Boyman O (2015) Interleukin-2: biology, design and application. Trends Immunol 36:763–777
Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, Votavova P, Pepper M, Kovar M, Garcia KC (2015) Antibodies to interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity 42:815–825
Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, Ju W, Fischer S, Oh J, Spolski R, Weiskopf K, Kohrt H, Foley JE, Rajagopalan S, Long EO, Fowler DH, Waldmann TA, Garcia KC, Leonard WJ (2015) Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity 42:826–838
Emerson SD, Palermo R, Liu CM, Tilley JW, Chen L, Danho W, Madison VS, Greeley DN, Ju G, Fry DC (2003) NMR characterization of interleukin-2 in complexes with the IL-2Ralpha receptor component, and with low molecular weight compounds that inhibit the IL-2/IL-Ralpha interaction. Protein Sci 12:811–822
Mott HR, Baines BS, Hall RM, Cooke RM, Driscoll PC, Weir MP, Campbell ID (1995) The solution structure of the F42A mutant of human interleukin 2. J Mol Biol 247:979–994
Sauvé K, Nachman M, Spence C, Bailon P, Campbell E, Tsien WH, Kondas JA, Hakimi J, Ju G (1991) Localization in human interleukin 2 of the binding site to the alpha chain (p55) of the interleukin 2 receptor. Proc Natl Acad Sci USA 88:4636–4640
Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, Pande VS, Fathman CG, Boyman O, Garcia KC (2012) Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 484:529–533
Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, Lang S, Roemmele M, Hofer T, van Puijenbroek E, Wittig D, Moser S, Ast O, Brünker P, Gorr IH, Neumann S, de Vera Mudry MC, Hinton H, Crameri F, Saro J, Evers S, Gerdes C, Bacac M, van Dongen G, Moessner E, Umaña P (2017) Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. OncoImmunology 6:e1277306
Carmenate T, Ortíz Y, Enamorado M, García-Martínez K, Avellanet J, Moreno E, Graça L, León K (2018) Blocking IL-2 signal in vivo with an IL-2 antagonist reduces tumor growth through the control of regulatory T cells. J Immunol 200(10):3475–3484
Bianchi M, Meng C, Ivashkiv LB (2000) Inhibition of IL-2-induced Jak-STAT signaling by glucocorticoids. Proc Natl Acad Sci USA 97:9573–9578
Acknowledgements
The author acknowledges utilization of the following simulation and visualization software packages: (1) NAMD and (2) VMD: NAMD and VMD, developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois, Urbana-Champaign. (3) Discovery Studio Visualizer: Discovery Studio Modeling Environment, Release 3.5, Accelrys Software Inc., San Diego, 2012.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material. Structure and Function of Interleukin-2 (PDF); Figs. S1–S5; Tables S1, S2.
Rights and permissions
About this article

Cite this article
Roy, U. Structure and Function of an Inflammatory Cytokine, Interleukin-2, Analyzed Using the Bioinformatic Approach. Protein J 38, 525–536 (2019). https://doi.org/10.1007/s10930-019-09833-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-019-09833-8