
Abstract
Herein, the preparation of polyurethane nanofibrous microfiltration membranes, with electrospinning and then dip-coating methods was reported. The study of process parameters (i.e. needle-free electrospinning and dip-coating condition) on the membrane properties was also conducted. The different pore sizes of the prepared MF membranes (e.g. 0.23, 0.33, and 0.47 μm) is orchestrated through the adjustment of various electrospinning and dip-coating parameters. The capability of these membranes to simultaneously eliminate the sources of water pollution, i.e. micro-particles and bacteria, has been demonstrated. Specifically, the prepared membranes could thoroughly reject the E. coli BL21 (DE3) bacteria (~ 97–99%) as well as micro-particles through size extrusion mechanism (~ 95–99%), while they retained a high permeation flux (~ 65,400, ~ 40,000 and ~ 25,700 (L/m2 h bar) for 0.46, 0.33, and 0.25 μm pore size, respectively). In addition, a comparison between nanofibrous MF membranes and their commercial counterparts from both utility and effectiveness standpoint was conducted and the obtained result indicated the supreme performance of these membranes in comparison with the commercial membranes of the same mean pore size, also with almost double or triple higher flux.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Increasing global population growth rate, industrialization, environmental pollution concerns, and the deficit of available resources, has made the issue of water treatment an emergent necessity [1, 2]. Therefore, developing a more practicable purification technique from both efficiency and cost aspects is indispensable [3]. Amongst the wastewater treatment technologies, pressure-driven membrane filtration processes e.g. microfiltration (MF), ultrafiltration (UF), nanofiltration (NF), and reverse osmosis (RO), have proven be high-efficient and markedly accessible [4]. In particular, MF membrane featuring pore sizes in the 0.1–10 μm range are able to fulfill the pretreatment of wastewater and the rejection of the most of bacteria as well as other contaminants from polluted water [5, 6]. Sterilization of the polluted water by means of MF membranes is highly favored since most of the micro-organisms in aqueous media involved and conveyed on the water are normally sized greater than 0.5 μm [7]. Hence, the majority of commercial membranes (CMs) such as Sartorius or Millipore, with nominal 0.22 μm and 0.45 μm pore sizes, were accustomed for MF utilizations.
Electrospun nanofibrous membranes (ENMs), as a state-of-the-art material suitable for MF application, could in a great effect prevail over the disadvantages of phase inversion procedure and dominate the conventional polymeric membranes prepared thereof. The ENMs offers profuse benefits including a lower basis weight, a larger surface area as well as a higher porosity and permeability [8]. The broad utilization of fibrous membranes in segregation applications lies in their properties to form a web-like and spider web-like interconnected structure of a high surface area which could entrap and adsorb various pollutant microorganisms. Electrospinning is a versatile method with extreme potential for producing a non-woven nanofibrous fabric of high interest in water purification techniques, i.e. NF [9], UF [10] and MF [11] which has recently been the center of variant research attentions.
There are two main strategies for preparation of controlled pore size nanofibrous microfiltration membranes: electrospinning condition and chemical or physical modification of nanofibrous membranes. Wang et al. reported the preparation of desired acrylonitrile (PAN) nanofibrous scaffold featuring narrow fiber diameter distribution and a favorable thickness for MF processes based on electrospinning parameters [8]. Also, Hsiao et al. suggested that polyvinylalcohol (PVA) nanofibrous membrane could be used as a MF membrane after the crosslinking with glutaraldehyde (GA) [12]. Kaur et al. demonstrated the utility of plasma-induced graft copolymerization of methacrylic acid monomer on poly (vinylidene fluoride) nanofibrous mats as a modification method for the preparation of MF membranes [13].
The needle and needle-free electrospinning strategies, as two widely accepted procedures of nanofibers mats production, have been vastly employed and developed. Up to the present, numerous researches have extensively made use of the lab-scale needle electrospinning method for the preparation of nanofibrous structured products. However, this method faces with constraints including low nanofiber producing rate, industrial applicability, needle blocking, and the non-continuity of the system. Hence, different needle-free techniques were progressed by several researchers to address these limitations [14,15,16]. Moreover, the exquisite characteristics of PU nanofibers involving: prodigious morphology, durable spinning susceptibility, and brilliant mechanical characteristics have attracted a great deal of attention through researchers for a diverse range of applications [17,18,19].
In consequence, the wiry needle-free electrospinning with two porous wiry systems and dip-coating techniques were the methods of choice in this study for producing the PU ENMs. The nanofibrous membrane pore sizes are principally resulted from interpenetration of highly interconnected nanofibers. Thus, the fiber diameter and its morphology have shown to be the main determinants of the mean pore size of nanofibrous samples which this has been addressed in a number of literatures [20,21,22].
The purpose of the current study was to prepare the high efficiency PU nanofibers MF membranes using a needle-free electrospinning strategy with high throughput. In this way, PU ENMs membranes preparation alongside the aim of study on optimization of needle-free electrospinning parameters such as electrospinning technique, polymer concentration, and additives types was conducted. The effect of these parameters on the pore size of the nanofibrous membranes was also investigated. Also, dip-coating method as another strategy was applied for decreasing of nanofibrous membranes pore size and the results of this method was investigated.
Experimental
Materials
The thermoplast elastomer PU (Elastollan®1180A10) was purchased from BASF Company. Non-woven poly (ethylene terephthalate) (PET) substrate was obtained from Sanko, Japan (No. 16-1) as a support layer or baking material (BM). Poly(vinyl alcohol) (PVA, Mw ~ 9000, 80% hydrolyzed), Glutaraldehyde (GA, 25% in H2O) was prepared from Sigma–Aldrich. Standard size micro-particles (polybead carboxylate) in the sizes of 1.0, 0.50 and 0.20 μm were purchased from Polysciences, Inc. Tetrahydrofuran (THF), Lithium chloride(LiCl), Tetraethylammonium bromide (TEAB), N,N-dimethylformamide (DMF, 99.8%), Ethyl alcohol (denatured alcohol) and E. coli BL21 (DE3) bacteria were obtained from Sigma–Aldrich. CMs in MF range (Model GSWP, Millipore) with the pore sizes of 0.22 μm and 0.45 μm were tested to be used in comparison objectives.
Fabrication of Nanofibrous Membranes
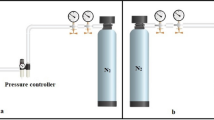
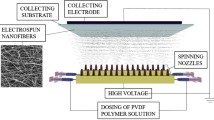
The home-made wiry needle-free electrospinning system was employed in this study for the fabrication of nanofibrous membranes as shown in Fig. 1a. It is composed of two porous wires with length of 500 mm and diameter of 2 mm connected to the positive pole of high voltage power supply, two polymeric solution holders (volume: 1 L) which can be filled with the identical or different concentrations of a polymer, a collector sheet with length of 650 mm, and the collector electrode connected to the negative section of high voltage system located at the end top of the wiry electrodes. When the polymeric solution holders scanned all of the wires, the polymeric solution is poised to initiate the coating and reloading the entire porous wiry nozzles. The holders and take-up cylinder speed for oscillating the PET support layer as a BM were adjusted at 50 mm/s and 2 m/h, respectively. PU solutions, in the concentration of (10 w/v% and 5 w/v%) were prepared by dissolving in (50/50 v/v) DMF/THF solvent ratio at room temperature. TEAB and LiCl in the concentration of 0.1 wt% and 0.04 wt%, respectively, were added as a polymeric solution modifier. The other needle-free electrospinning para meters were outlined in Table1.

Schematic design of the materials processing systems
Modification of PU Nanofibrous Surface
After optimizing of the electrospinning conditions, the pore size of nanofibers membranes is reduced by dip-coating modification. For this end, PU nanofibrous membranes were immersed in the solution of polyvinylalcohol (0.1 w/v% concentration in water) and glutaraldehyde (0.001 w/v% in water) for different time. In this process, the coating process occurs in PVA/GA dilute solution in the PU nanofibrous network and as a result the pore size of membranes was reduces. After coating process, the residual polymeric solution was blotted from the nanofibrous membrane surface and then rinsed with deionized water for three times. At the end, the membranes were dried at room temperature. So, the MF membrane was obtained for practical filtration test (Fig. 1b).
Capillary Flow Porometry
The mean flow pore size (average pore size) and largest pore size (bubble-point) of the PU nanofibrous membranes were measured with home-made porometer (capillary flow porometer, ASTM-F316-03) depicted in Fig. 1c (left) [23]. The operating procedure of capillary flow porometer is as follows: In the first step, an inert gas, e.g. N2 flows within a dry membrane and then from the same membrane after which has been soaked by wetter liquid. Herein, ethyl alcohol possessing a certain surface tension of 22.3 dynes/cm was utilized as the wetter liquid agent [23]. Then in the next step, the diagrams of gas flows versus pressures are plotted for dry and wet samples.
After drawing of wet flow, dry flow, and half-dry flow as a porometry characterization for membrane, the method through which the largest and average pore size (mean flow pore size) of these curves are determined is as follows. The porosity, in the wet flow, initiates from the largest (i.e. bubble point) towards the smallest one by the time which the pressure of N2 gas is elevated. Hence, the pressure at which the wet flow starts to increase is attributed to the largest pore and that of the half-dry gas flow and wet flow intersection is related to the mean flow pore size. The largest and mean flow pore sizes of membranes were calculated through the mentioned pressure points in the plotted curves utilizing the pressure-pore size formula Eq. 1 [23]:
In which D represent the pore diameter membrane (μm); θ represent the contact angle of the standard wetter agent; p represents the pressure (Pa); γ represents the surface tension of the wetter agent (dyn/cm). For this measurement, the membranes were cut into round circular shape with 4 cm diameter.
Microfiltration Performance
A home-made dead-end filtration system was employed so as to measure the pure water flux as well as micro-particle retention ratio of MF membranes, which is schematically depicted in Fig. 1c (right). The membranes were cropped in the forms of round circular shapes with a diameter of 4 cm and all of the chosen membranes owned a total thickness of ~ 175 ± 11 μm and that for the electrospun PU layer was about 75 μm. The pure water permeation is generally representative of the necessary energy for filtration and is a practical test method for the permeability comparison of various membranes. For this test, all of the nanofibrous and commercial MF membranes were perfectly soaked and subsequently been examined under different pressures ranging from 0.07 to 2.77 bar. The method of the study for assessing the micro-particle rejection ratio of the MF membranes was due to the following procedure. At first, aqueous suspensions of polybead carboxylate micro-particle (100 ppm) (0.20, 0.50 and 1.0 μm) with microsphere morphology were prepared via ultrasonic dispersion method. In addition, before the test, all of the membranes pre-compressed at 1 bar and then a 10 mL suspension of micro-particles was filtered from the nanofibrous and CMs with dead-end separation set-up. For measurement of the carbon concentrations in the feed and the permeate solutions, the total organic carbon analyzer (TOC, Shimadzu TOC-VCPN) was applied. The rejection/retention ratio of the standard-sized particles was calculable according to the Eq. 2:
where Cf and Cp displays the concentration of organic carbons in the feed and permeate solution, respectively. To exclude the precision concerns, the reported results are the average of three times repetition. A dead-end filtration system equipped with 5 mL of E. coli BL21 (DE3, 106 CFU/mL) (Micro-syringe, Filter Holder 25 mm, Millipore) was applied for the bacteria rejection test purposes. The sterilization of the membranes as well as all of the filtration equipment pieces including cells was accomplished using gamma ray (γ-ray) and autoclave at 121 °C for 20 min, respectively. The bacteria rejection/retention evaluation was carried out at ambient conditions and under a steady flow rate in which a model contaminant compound was utilized to monitor the bacteria growth. The concentration of bacterial in the feed and permeate solutions and also the terms of LRV (Log Reduction Value) was calculated through the colony assay method. The latter was applied to calculate the filtration performance and bacterial retention ratio according to Eq. 3:
where Cf and Cp express the bacterial concentrations in the feed and permeate solutions (CFU/mL), respectively.
The porosity of ENMs as a function of packing density was calculated using the Eq. 4 [24]:
To determine the former, membrane of about 2 cm2 and 175 ± 11 μm were weighted for apparent density (ρ′) and also the bulk density (ρ) of PU granule was defined to be 1.11 g/cm3. The porosity of the membranes was estimated from the average of three membranes. The micrometer was used to detect the total thickness of the membranes (i.e. baking material and electrospun layer).
Scanning Electron Microscopy (SEM)
The morphological studies and fiber orientation of the all nanofibrous membranes were studied using scanning electron microscopy (SEM, JEOL, JSM-5600LV). The average nanofiber diameter of PU nanofibrous mats were detected through the SEM image analyzer software. All membranes were coated by gold for 35 s prior to the SEM analysis.
Results and Discussion
Morphological Study of PU Nanofibrous Membranes
Study on the morphological characteristics of ENMs produced via needle-free electrospinning method was accomplished through SEM. Firstly, both polymeric solution holders 1 and 2 has been filled with the same concentration and thereafter the electrospinning was started throughout the two wiry-nozzles. Figure 2a illustrates the SEM images of the electrospun membrane obtained using 10 w/v% PU in 50/50 v/v DMF/THF named M1 herein. The fiber diameter of the membrane was 195 ± 15 nm with an inhomogeneous distribution and architecture. Furthermore, some areas including film-fiber structure, beaded-fibers area, and beads could be noticed in the membrane structure. One of the foremost significant parameters affecting the nanofiber diameter is the polymeric concentration which facilitates the preparation of membranes of smaller pore sizes and diameters. Therefore, lower polymer concentrations were used to obtain nanofibrous membranes with smaller fiber diameter. Per contra, the decrease in polymer concentration value has resulted in the increment of the surface tension of the polymeric solution. This results in unfledged elongation of the nanofibrous jets in the electrical field and also in the formation of beads, beaded-fibers, and film-fiber zones [25]. The defective morphology caused by these improper structures had led to the membrane porosity reduction. To overcome this obstacle and improve nanofibrous mats properties, various strategies have been employed such as additives utilization, polymer concentration alteration, electrical field applying, and a mixture of solvents [26, 27]. As stated earlier, additives of LiCl and TEAB, as well as variant low polymer concentration, were applied for the improvement and preparation of uniform nanofibrous structure and also, for dip-coating method, different time was applied in this work tabulated in Table 2.

SEM surface architectures of M1 (a) and M2 (b) nanofibrous membranes and schematic of chemical interaction between TEAB and polyurethane chains (c)
Figure 2b shows SEM images of the produced electrospun membrane using 10 w/v% PU in 50/50 v/v DMF/THF in the presence of 0.04%wt LiCl (M2). It is obvious that adding LiCl as an additive has assisted to the preparation of the PU nanofibrous mat with a reduced fiber diameter of 220 ± 15-nm and narrow-size distribution. In addition, the beaded and fiber-film area were eliminated which enhanced the uniform regularity of the prepared nanofibrous mat. Basically, the electrical conductivity of polymeric solution was facilitated through the addition of LiCl salt [28]. Furthermore, PU shows a negative or positive surface charge depending on its medium, however, the higher density of amine groups in the PU structure makes it predominantly act as a Lewis base [29, 30]. Given this, the surface charge of PU in the polymeric solution is intensified through Lewis acid–base interaction with LiCl, and the surface charge of polymeric chains is further increased. Consequently, as a secondary effect of LiCl addition, the applied electrical force of high voltage sorce to the polymericc chains is elevated. Hence, the electrical conductivity improvement of polymeric solution as well as the elongation of the polymeric chains and nanofibrous jets with higher forces under the same electrical field were the outcomes of LiCl attendance. As a result, the electrical force could overcome the surface tension and the nanofibrous mat possessing highly uniform structure and porosity (~ 87%) was obtained [28, 31, 32].
The mechanism of TEAB performance as an additive in polymeric solutions is followed through three main strategies depending on the utilized concentration. Provided that the lower concentration of TEAB was selected it would have been acted as a salt responsible for electrical conductivity intensifying of the polymeric solution which, in turn, has resulted in higher elongation force exertion from electrical fields. As a result of polymeric jets elongation, the beaded-nanofiber formations would have been decreased. That is to say, a higher electrical force as a result of higher electrical conductivity could overcome the surface tension of polymeric solution to a great effect and led to the formation of a nanofibrous membrane of narrower fiber diameter and a stable uniform nanofibrous jet architecture. Conversely, providing that the higher concentration of TEAB was applied it would have been acted as a binder that interacts between this additive and polymeric chains in the solution and further caused to diameter increment of nanofibrous mats. This could elevate the PU chains polarity in electrical field and which leads to its co-adsorption with an electrostatic force and subsequent enhancement of the polymeric chains number and of the polymeric concentration in nanofibrous jets. Thereupon, the diameter of jets was increased which resulted in the nanofiber diameter enhancement [33]. The schematic mechanism of the aforementioned interaction is depicted in Fig. 2c.
The medium concentration of TEAB not only decreases the nanofiber diameter but also leads to a higher interconnected nanofibrous network. Figure 3a represents SEM images of the nanofibrous membrane fabricated using 10 w/v% of PU in a medium consisting of equal proportion (50/50) of DMF/THF and 0.1 wt% TEAB as an additive in the solvent mixture. The bead-free, sizeable, open pore, and slick skeleton of high porosity (~ 85%) of M3 could perspicuously been observed. Also, the diameter of nanofiber and porosity of M3 are lower than that of M2 (180 ± 20 nm, ~ 85% and 220 ± 15 nm, ~ 87% for PU/TEAB and PU/LiCl, respectively). According to SEM images, the PU-TEAB (M3) nanofibrous membrane is of a different morphology compared to PU-LiCl (M2) membrane. The density of interaction is proper in the medium concentration causing the interconnection of nanofiber in the intersection area. Consequently, the proper concentration of this additive brings about lower fiber diameter and higher interconnection.

SEM surface architectures of M3 (a), M4 (b) M5 (c) M6 and M7 (d), M8 (e), M9 (f), M10 (g) and M11 (h) nanofibrous membranes
In the second step, to produce a nanofibrous membrane with smaller fiber diameter and pore size, the differential concentration technique, i.e. changing the polymer concentration in during process, was utilized. In this method, initially, the polymer with a higher concentration is electrospun by shifting the substrate to the front, afterward the polymer with different concentrations and properties. As a result, the resulting nanofibrous membrane have a three layers including (i) sub-layer (a non-woven PET, a backing material), (ii) mid-layer (nanofiber with with 10%w/v PU in 50/50 DMF/THF and 0.04%wt LiCl) and (iii) top-layer (nanofiber with concentration of 5%w/v in a medium of 50/50 DMF/THF and 0.04% wt LiCl as an additive in the solvent mixture). Figure 3b shows SEM images of electrospun nanofibrous membrane produced using the aforementioned structure. The images illustrate that with the decrement of polymer concentration down to 5% the fiber diameter decreased from 220 ± 15 to 138 ± 10 nm. From images, it is clear that the morphology of M4 sample is different from M2. Reducing the concentration of polymer not only resulted in the nanofiber diameter reduction and porosity decreasing to ~ 83% but also in the enhancement of nanofibers density.
SEM images of M5 nanofibrous MF membrane is shown in Fig. 3c. In this case, the holders 1 and 2 were filled by the polymeric solution in the concentration of 10% w/v of PU in the equal proportion of 50/50 DMF/THF and 0.1%wt TEAB as well as 5%w/v PU in 50/50 DMF/THF with 0.1%wt TEAB in the solvent mixture, respectively. The images demonstrate that with the decrement of polymer concentration down to 5% the nanofiber diameter decreased from 180 ± 20 to 118 ± 5 nm. Furthermore, the morphology (i.e. interconnecting, fiber density and pore sizes) of M5 is differing from other nanofibrous membranes as well as M4 and the porosity of this nanofibrous MF membrane (~ 80%) is lower compared to the earlier mentioned membrane.
In this way, to further reduce the membrane pore size, the polymeric concentration was altered. For M6, the holders 1 and 2 were filled from polymeric solution in the concentration of 10% w/v PU in the medium of 50/50 DMF/THF with 0.04% wt LiCl and 3%w/v PU in the medium of 50/50 DMF/THF with 0.04% wt LiCl, respectively. For M7, the same concentrations were used with 0.1% wt TEAB as an additive. As the Fig. 3d (M7 in the left and M8 in the right) indicates, by reducing the concentration of polymer from 5 to 3% in the Holder 2, in addition to the nanofibrous structure, the film structures also appear and proper electrospinning properties at this concentration have not been observed. Dropping down the concentration of polymer has resulted in the increment of solvent content in the solution and thus in the nanofibrous jets. Heretofore, the co-deposition of wet nanofibers on each other and on BM is occurred and nanofiber-film structure is created. On the other hand, the surface tension increases with this concentration in solution. Hence, the nanofibrous jets were not adequately elongated in the electric field which caused in the formation of beaded, film-fiber structure, and reduction of porosity of M6 and M7 (73% and 70% for M6 and M7, respectively). As a supposition, in the lower polymeric concentration, while the other conditions are constant, the membranes would have been inclined to form an unconventional and non-uniform film structure which is not desired for MF applications.
As shown in M6 and M7 samples, the pore size of nanofibrous membranes can’t be reduced by changing the concentration and electrospinning condition. Because of film-fiber and beaded structure, a uniform membrane will not be produced during electrospinning. Therefore, a second strategy (dip-coating) was used to modify and reduce the size of the membrane cavities. For this end, the M2 sample was selected as based membrane for modification by this method. Figure 3e shows the SEM analysis of M8 produced by 20 s coating time in PVA/GA solution. As can be seen from the images, no specific changes in the morphology of nanofibrous membranes have been occurred. Therefore, it can be concluded that no change in membrane pore size has happened.
In the following of coating process, by increment of coating time to 60 s, the morphology of nanofibrous membranes surface was changed. As shown in Fig. 3f, the diameter of nanofiber is thickened and the distances among nanofibers was coated by cross-linked PVA/GA in nanofibrous network. It is also quite clear that the porosity of M9 is reduced compared to the M8 or M2 samples (87% and 78% for M2 and M9, respectively). Therefore, it can be concluded that the pore size of the membrane has decreased.
Figure 3g demonstrates the SEM analysis of membrane prepared through 120 s coating time. In a comparative point of view, in the higher coating time, the membrane porosity (74%) and interface distance among nanofibers were apparently more reduced. Also, in the higher coating time (M10 in this case), the pore sizes of membranes were obviously decreased which is the representative of the PVA/GA dip-coating progress.
In higher dip-coating time (180 s) with the same PVA/GA concentration, there is tendency for the membrane surface to form film structure which is not desirable for MF proccess. From Fig. 3h, M11, increasing of coating time has resulted to forming a complete and undesirable film throughout the membrane surface which makes it unsuitable for the MF applications. Also, it can be seen that the morphology of the nanofibers is not clearly seen. So as aresult, the optimum dip-coating time and concentration should be considered. According to the result of SEM analysis, the pore size of the M2, M3, M4, M5, M9 and M10 was investigated.
Porometry Evaluation of PU Nanofibrous Membranes
For characterizing the MF membranes pore size, a hand-made porometer system (capillary flow porometery, ASTM-F316-03) was employed in the current work. Denatured alcohol with a determined surface tension (22.3 dynes/cm) was applied as the wetter liquid for pore size analysis. Figure 4 illustrates the dry, wet, and half-dry flow schemes for PU MF membranes and the membrane’s pore characterizations were detected from these diagrams. Figure 4a(I) is related to the diagram data of M2 ((10 w/v% concentration of PU in equal 50/50 v/v DMF/THF and 0.04%wt of LiCl additive in the solvent mixture). The pressure of the starting point of the wet flow is located at 0.16 bar and the largest estimated pore through the supposed pore-pressure formula [Eq. (1)] was 4 microm. The pressure attributing to mean flow pore size was 0.75 bar and the associated pore diameter was 0.85 μm. Because of the decrement of fiber diameter and porosity as well as the increment of fiber density, the bubble point and mean flow pore size of M3 (10 w/v% PU, 50/50 DMF/THF medium and 0.1 wt% TEAB), has been decreased as shown in Fig. 4a(II) The bubble point pressure of M3 was 0.18 bar and the bubble point of this sample was 3.4 μm and the mean flow pore size pressure happened at 0.81 bar and the related mean flow pore size was detected to be 0.78 μm. As the polymeric concentration was decreased, the largest pore and average pore size has grown smaller scale. Figure 4a(III) represents the porometry behavior of M4 (5%w/v of PU in 50/50 v/v DMF/THF mixture solvent and 0.04% wt LiCl). As deducted from the images, the pressure correlated to largest pore was 0.21 bar and the bubble point of this membrane was 2.9 μm. The pressure related to intersection of the wet and half-dry flow was 1.2 bar and the mean flow pore size for M4 was 0.52 μm. Mitigating the polymeric concentration led to the fiber diameter decreasing and as a result, a membrane with smaller pore size was prepared. Figure 4a(IV) illustrates the porometry characterization for M5 sample. The largest and mean pore size has become decreased in which the former was occurred at 0.3 bar and the latter was 2.1 μm in size for the largest one. In addition, the intersection of wet and half-dry flow was located at 1.35 bar and the corresponded mean pore size was 0.47 μm. Figure 4a(V) is related to M9 produced by 60 s dip-coating time. As seen from image, the pressure correlated to bubble point was 0.66 bar and the largest pore size of this sample was 0.96 μm. The pressure related to intersection of the wet and half-dry flow was 2.12 bar and the mean flow pore size for M9 was 0.33 μm. By increasing of dip-coating time to 120 s the mean flow pore size and bubble point were more decreased. Figure 4a(VI) shows the porometry characterization of M10. For this membrane the pressure of bubble point was occurred at 1.04 bar and the largest pore size was 0.61 μm. Also, the intersection of wet and half-dry flow was set at 2.8 bar and the related mean pore size was 0.23 μm. To derive a deduction from the findings, changing the electrospinning and dip-coating condition might result in the largest and mean flow pore size decrement. Accordingly, different nanofibrous MF membranes of various pore sizes were prepared. Regarding that most commercial MF membranes are in the size of 0.22–0.45 μm, the separation performance of M5, M9 and M10 samples as a MF membranes featuring 0.47 μm, 0.33 μm and 0.23 μm pore sizes, respectively, were evaluated and compared with their commercial competitors.

The pore-size, water flux and micro-particles separation analysis results
Microfiltration Evaluation
The pure water flux, micro-particle, and bacteria rejection/retention of PU ENMs (M5, M9 and M10 with pore sizes of 0.47 μm, 0.33 μm and 0.23 μm, respectively) were examined so as to evaluate the capability of electrospun PU membranes possessing an overall thickness of 175 ± 11 μm to operate as a MF filter. Also, to bring a comparative view, their results along with those of CMs with 0.22 μm and 0.45 μm pore size and ~ 180 μm thickness, produced for MF usages, were plotted in Fig. 4b. Prior to the experiment, all nanofibrous and commercial MF membranes were flushed at 1 bar in water for 10 min to obviate the challenges of all shut-in air pocket and the membranes pores blockage. The pure water flux vs. pressure experiment approved the superiority of ENMs over CMs as shown in Fig. 4b. The interconnected nanofibrous network, high porosity and open pore structure of PU nanofibrous MF membranes have made for about double/triple higher flux in comparison with CMs over a certain pressures range. The result showed that holding the water influx pressure low, for nanofibrous MF membranes desirable in low-pressure filtration application, was quite indispensable.
The standard-sized micro-particles (polybead carboxylate) were employed as the test indicators in evaluating the MF efficiency of the ENMs. Since the polybeads are of spherical morphology, their utilization could be a criterion for ENMs and CMs pores size scales. As shown in Fig. 4c, the nanofibrous membranes, as well as the conventional membranes, represented almost identical rejection ratio on 0.2 μm, 0.50 μm and 1.0 μm standard-sizes particles. Hence, the mechanism of rejection for these MF membranes could be explained as follows. Due to the mean pore size similarity of the PU ENMs and CMs, both demonstrate a quite analogous filtration performance in standard particles retention and thus the process of microspheres treatment might be justified by sieving effect or size exclusion.
The bacteria rejection was performed on the PU ENMs and CMs (total thickness of nanofibrous and CMs is 175 ± 11 μm and ~ 180 μm, respectively). 10 mL E. coli BL21 (DE3) bacteria (106 CFU/mL) aqueous suspension was applied as the blank suspension for monitoring the bacteria growth. As regards the larger size of the bacteria than that of the mean flow pore of all CMs and ENMs, the bacteria were entirely omitted and there was no trace of E. coli BL21(DE3) bacteria detectable throughout the permeate solution. Also, LRV of ~ 6 was obtained for all of the nanofibrous and commercial MF membrane (Table 3).
Conclusion
The highly efficient wiry needle-free electrospinning technique was utilized to fabricate of PU electrospun MF membranes. It was implied that this method has prominently made great strides in the production rate of PU nanofibers and has higher yields than that of traditional stand-alone needle electrospinning technique. The effects of electrospinning parameters on corresponding pore sizes was systematically investigated. In the following, for further decrement of pore size, dip-coating method was applied. In this method, the effect of modification time was evaluated. Ultimately, a comparative study between nanofibrous MF and CMs on the MF performance i.e. their functionality in removing microparticles and bacteria was conducted and result indicated that nanofibrous MF membrane rendered remarkably higher flux performance (2–3 times) over the Millipore microfiltration membranes having the same mean flow pore size. Moreover, they retained a high rejection rate in the entire micro-particle retention examinations and could realize the ideal state of bacteria removal.
References
Gleick PH (1998) The human right to water. Water Policy 1:487–503. https://doi.org/10.1016/S1366-7017(99)00008-2
Cheng H, Hu Y, Zhao J (2009) Meeting China’s water shortage crisis: current practices and challenges. Environ Sci Technol 43:240–244. https://doi.org/10.1021/es801934a
Quist-Jensen CA, Macedonio F, Drioli E (2015) Membrane technology for water production in agriculture: desalination and wastewater reuse. Desalination 364:17–32. https://doi.org/10.1016/j.desal.2015.03.001
Reith C, Birkenhead B (1998) Membranes enabling the affordable and cost effective reuse of wastewater as an alternative water source. Desalination 117:203–210. https://doi.org/10.1016/S0011-9164(98)00097-6
Dobrowsky PH, Lombard M, Cloete WJ, Saayman M, Cloete TE, Carstens M, Khan S, Khan W (2015) Efficiency of microfiltration systems for the removal of bacterial and viral contaminants from surface and rainwater. Water, Air, Soil Pollut 226:33. https://doi.org/10.1007/s11270-015-2317-6
Fernández García L, Álvarez Blanco S, Riera Rodríguez FA (2013) Microfiltration applied to dairy streams: removal of bacteria. J Sci Food Agric 93:187–196. https://doi.org/10.1002/jsfa.5935
Wang Y, Hammes F, Düggelin M, Egli Th (2008) Influence of size, shape, and flexibility on bacterial passage through micropore membrane filters. Environ Sci Technol 42:6749–6754. https://doi.org/10.1021/es800720n
Wang R, Liu Y, Li B, Hsiao BS, Chu B (2012) Electrospun nanofibrous membranes for high flux microfiltration. J Membr Sci 392:167–174. https://doi.org/10.1016/j.memsci.2011.12.019
Mahdavi H, Moslehi M (2016) A new thin film composite nanofiltration membrane based on PET nanofiber support and polyamide top layer: preparation and characterization. J Polym Res 23:257. https://doi.org/10.1007/s10965-016-1157-4
Yang Y, Li X, Shen L, Wang X, Hsiao BS (2017) Ionic cross-linked poly (acrylonitrile-co-acrylic acid)/polyacrylonitrile thin film nanofibrous composite membrane with high ultrafiltration performance. Ind Eng Chem Res 56:3077–3090. https://doi.org/10.1021/acs.iecr.7b00244
Moslehi M, Mahdavi M (2019) Controlled pore size nanofibrous microfiltration membrane via multi-step interfacial polymerization: preparation and characterization. Sep Purif Technol. https://doi.org/10.1016/j.seppur.2019.04.041
Liu Y, Wang R, Ma H, Hsiao BS, Chu B (2013) High-flux microfiltration filters based on electrospun polyvinylalcohol nanofibrous membranes. Polymer 54:548–556. https://doi.org/10.1016/j.polymer.2012.11.064
Kaur S, Ma Z, Gopal R, Singh G, Ramakrishna S, Matsuura T (2007) Plasma-induced graft copolymerization of poly (methacrylic acid) on electrospun poly (vinylidene fluoride) nanofiber membrane. Langmuir 23:13085–13092. https://doi.org/10.1021/la701329r
Wang L, Zhang Ch, Gao F, Pan G (2016) Needleless electrospinning for scaled-up production of ultrafine chitosan hybrid nanofibers used for air filtration. RSC Adv 6:105988–105995. https://doi.org/10.1039/C6RA24557A
Yarin AL, Zussman E (2004) Upward needleless electrospinning of multiple nanofibers. Polymer 45:2977–2980. https://doi.org/10.1016/j.polymer.2004.02.066
Dosunmu OO, George GG, Kataphinan W, Reneker DH (2006) Electrospinning of polymer nanofibres from multiple jets on a porous tubular surface. Nanotechnology 17:1123–1127. https://doi.org/10.1088/0957-4484/17/4/046
Ge JC, Choi NJ (2017) Fabrication of functional polyurethane/rare earth nanocomposite membranes by electrospinning and its VOCs absorption capacity from air. Nanomaterials. 7:60. https://doi.org/10.3390/nano7030060
Erdem R, Usta I, Akalin M, Atak O, Yuksek M, Pars A (2015) The impact of solvent type and mixing ratios of solvents on the properties of polyurethane based electrospun nanofibers. Appl Surf Sci 334:227–230. https://doi.org/10.1016/j.apsusc.2014.10.123
Gorji M, Jeddi AAA, Gharehaghaji AA (2012) Fabrication and characterization of polyurethane electrospun nanofiber membranes for protective clothing applications. J Appl Polym Sci 125:4135–4141. https://doi.org/10.1002/app.36611
Ryu YJ, Kim HY, Lee KH, Park HCh, Lee DR (2003) Transport properties of electrospun nylon 6 nonwoven mats. Eur Polym J 39:1883–1889. https://doi.org/10.1016/S0014-3057(03)00096-X
Li D, Frey MW, Joo YL (2006) Characterization of nanofibrous membranes with capillary flow porometry. J Membr Sci 286:104–114. https://doi.org/10.1016/j.memsci.2006.09.020
Eichhorn SJ, Sampson WW (2005) Statistical geometry of pores and statistics of porous nanofibrous assemblies. J. R. Soc. Interface 2:309–318. https://doi.org/10.1098/rsif.2005.0039
American Society for Testing and Materials (2003) Standard test methods for pore size characteristics of membrane filters by bubble point and mean flow pore test. ASTM, West Conshohocken, PA
Ma Z, Kotaki M, Yong Th, He W, Ramakrishna S (2005) Surface engineering of electrospun polyethylene terephthalate (PET) nanofibers towards development of a new material for blood vessel engineering. Biomaterials 26:2527–2536. https://doi.org/10.1016/j.biomaterials.2004.07.026
Huan S, Liu G, Han G, Cheng W, Fu Z, Wu Q, Wang Q (2015) Effect of experimental parameters on morphological, mechanical and hydrophobic properties of electrospun polystyrene fibers. Materials 8:2718–2734. https://doi.org/10.3390/ma8052718
Tan SH, Inai R, Kotaki M, Ramakrishna S (2005) Systematic parameter study for ultra-fine fiber fabrication via electrospinning process. Polymer 46:6128–6134. https://doi.org/10.1016/j.polymer.2005.05.068
Tang Z, Qiu Ch, McCutcheon JR, Yoon K, Ma H, Fang D, Lee E, Kopp C, Hsiao BS, Chu B (2009) Design and fabrication of electrospun polyethersulfone nanofibrous scaffold for high-flux nanofiltration membranes. J Polym Sci Part B Polym Phys 47:2288–2300. https://doi.org/10.1002/polb.21831
Yuan L, Wang R, Tian M, Qiu Ch, Fane AG (2013) Fabrication of polyvinylidene fluoride (PVDF) nanofiber membranes by electro-spinning for direct contact membrane distillation. J Membr Sci 425:30–39. https://doi.org/10.1016/j.memsci.2012.09.023
Cherukupally P, Sun W, Wong APY, Williams DR, Ozin GA, Bilton AM, Park CB (2020) Surface-engineered sponges for recovery of crude oil microdroplets from wastewater. Nat Sustain 3:136–143. https://doi.org/10.1038/s41893-019-0446-4
Cherukupally P, Acosta EJ, Hinestroza JP, Bilton AM, Park CB (2017) Acid−base polymeric foams for the adsorption of micro-oil droplets from industrial effluents. Environ Sci Technol 51:8552–8560. https://doi.org/10.1021/acs.est.7b01255
Liu Y, He JH, Yu JY, Zeng HM (2008) Controlling numbers and sizes of beads in electrospun nanofibers. Polymer Int 57:632–636. https://doi.org/10.1002/pi.2387
Qin XH, Wan YQ, He JH, Zhang J, Yu JY, Wang ShY (2004) Effect of LiCl on electrospinning of PAN polymer solution: theoretical analysis and experimental verification. Polymer 45:6409–6413. https://doi.org/10.1016/j.polymer.2004.06.031
Cengiz-Çallıoǧlu F, Jirsak O, Dayik M (2013) Investigation into the relationships between independent and dependent parameters in roller electrospinning of polyurethane. Text Res J 83:718–729. https://doi.org/10.1177/0040517512447587
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Moslehi, M., Mahdavi, H. Preparation and Characterization of Electrospun Polyurethane Nanofibrous Microfiltration Membrane. J Polym Environ 28, 2691–2701 (2020). https://doi.org/10.1007/s10924-020-01801-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-020-01801-z