
Abstract
Development of cotton fabric (CF) properties using nanocomposites via coating method was of considerable interest for wide applications. This article aims at developing CF properties by coating treatment using ethylene–vinyl-acetate (EVA), silicon dioxide (SiO2), aluminum oxide (Al2O3) nanoparticles and γ-irradiation widely used in waterproof and flame retardant applications. EVA-based nanocomposites, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3, were synthesized by γ-irradiation and the highest gel content of 81.2–95.3% was achieved at 30 kGy. The physicochemical properties of EVA-based nanocomposites were characterized by FT-IR, XRD, DSC and SEM techniques. Usage of irradiated EVA and EVA-based nanocomposites for treatment of CF by coating technique was successfully achieved. This technique provides a simple and versatile method leading to excellent uniform and smooth surface morphology without aggregation. The weight gain, mechanical properties, thermal properties, water vapor permeability and flame-retardant properties of the modified CF were evaluated. Moreover, compared with control CF, the resistivity of water absorptivity and hydrophobic property and the thermal stability were gained. The flame retardant properties of CF samples were performed using limited oxygen index (LOI) and vertical burning flame tests. LOI percentages of CF/EVA/SiO2, CF/EVA/Al2O3 and CF/EVA/SiO2/Al2O3 increased to 25.3, 27.5, and 29.3%, respectively. Untreated CF ignited and burned rapidly after 5 s. Meanwhile, the treated CF hold flame resistance properties and the burning time prolonged to 25 s. The results of the treated CF providing revealed hydrophobic and protective capability of the fabrics from being destroyed by burning, and support their further use in waterproof and flame retardant applications of fabrics.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Ethylene vinyl acetate (EVA) is a versatile commercial polymer material composed of nonpolar crystalline polyethylene and polar polyvinyl acetate segments that can vary greatly in their chemical and physical properties by the variation of the vinyl acetate content usually varies from 10 to 40%. The variation of the vinyl acetate content in EVA could alter many properties of EVA, such as degree of crystallization, glass transition temperature, thermal stability, and mechanical strength [1]. EVA is an elastomeric polymer that produces rubber-like materials. The material has gloss and good clarity, hot-melt adhesive waterproof properties, resistance to UV radiation, stress-crack resistance, and low-temperature toughness. Hence, EVA copolymers have a broad range of applications early in hot melt adhesives [2] coatings [3] wire and cable applications [4], footwear industry [5], controlled release drug systems [6], packaging applications [7], wearable electronic devices for human health monitoring [8]. Despite the promising aspects of EVA for coating applications, it has some disadvantages such as low tensile strength and highly flammable and produces large amounts of toxic smoke during combustion. In order to solve these problems, one of the investigated methods commits to incorporate flame retardants into the EVA composite materials to improve its properties.
Recently, metal oxide nanoparticles embedded within the polymer matrix, metal oxide-based polymeric nanocomposites, have attracted the growing interest in nanostructured materials due to the unique properties and combine the properties of the polymer as well as the metal oxide. Different types of metal oxide-based polymeric nanocomposites could be synthesized by different polymerization methods including suspension polymerization [9], emulsion polymerization [10], and γ-irradiation polymerization [11,12,13,14,15]. The properties of the produced nanocomposites depend on numerous aspects such as the morphological structure, the procedure of preparation, the nature of metal oxide nanoparticles that incorporated in, the structural features of the polymer used, and the chemical properties of both metal oxide and polymer molecules. The physicochemical properties of the nano-sized particles within the hybrid material differ markedly from those of molecular and bulk materials.
Silicon dioxide is one of the reinforced filling materials that are widely utilized as an improving agent in thermoplastic polymers that improves the mechanical properties [16] and has synergistic flame retardant property [17, 18]. SiO2 is known as a potential and safe flame retardant additive for polymer systems due to its high heat resistance, inert and non-toxic features [19]. SiO2 consists of silanol groups (Si–OH) which play a basic role in forming interaction with polar groups of polymers [20]. It is usually used as an enhancing agent in thermoplastic polymers to enhance the mechanical properties [16].
Alumina or aluminum oxide (Al2O3) nanoparticles are nano-sized particles from an alumina bulk. Al2O3 nanoparticles can be acquired by numerous methods including, sol–gel, pyrolysis, laser ablation, and sputtering [21]. Al2O3 has many inherent characteristics such as high melting temperature, low thermal expansion coefficient, toughness, hardness, non-toxicity, chemical inertness, transparency, and excellent insulating properties which make it advantageous as a nano-filler for polymer nanocomposites. Lately, several industrial applications of Al2O3 nanoparticles have been disseminated for example; drilling fluids agents [22], separators for lithium batteries [23], biosensors for vitamin E [24], enhancement of the thermal and mechanical properties of wood fibers [25]. Al2O3 has been extensively considered to serve as flame retardants, absorbents, catalysts, composite materials, and surface protective coatings [26]. Additionally, Al2O3 shows a good wear resistance and has a large surface area, so it is generally used as a filter for polymers to improve mechanical properties, catalyst, and catalyst support [27].
In this regard, γ-irradiation remains one of the most common processes that are capable of modifying the polymer-metal nanocomposites’ physical properties, which amplifies their utility in several applications [28, 29]. Notably, these alterations are attributed to the crosslinking or chemical bond scissions caused by high energetic radiation. This is because the ionization and excitation of the material’s molecules occurring during the interaction of ionization radiation with a polymeric material [30]. The radiation processing technique has numerous advantageous features including easy process control, simultaneous crosslinking of polymers and sterilization at the same time, and the technology is environmentally-friendly and no residue or pollutant in the surrounding environment, no additional chemical initiators or crosslinkers [31, 32].
Nowadays, functional materials, mainly flame-retardant textiles, have attracted much consideration besides an increase in the public awareness of safety. Worldwide, a large number of people die and lose fortunes in fire accidents due to the easy ignition of textiles every year [33]. Therefore, it is urgent to impart textiles with less flammability or non-flammability. In the last decades, numerous researchers have focused on the investigation of flame-retardant textiles and various preparation methods have been reported [34]. Cellulosic textiles, such as cotton fabric (CF), have been commonly utilized to produce industrial products, home furnishings, and apparel as a result of their advantages of pollution-free, vast quantities, strong biodegradability, and excellent mechanical properties [35].
There are countless combinations of coatings and fabrications methods for cotton fabrics to achieve different specifications for a wide spectrum of applications such as fabrication of reduced graphene oxide/silver nanoparticles onto cotton fabric for high electrical conductivity and superior electromagnetic shielding efficiency [36], the fabrication of highly durable electronic fabric for flexible electromagnetic performance via a facile dipping and drying method. which can address long term exposure towards the extreme environmental conditions both wet and dry media [37], fabrication of alkyl ammonium functional silsesquioxane /phytic acid complex as flame-retardant layer and hierarchical-structured titanium oxide@polydimethylsiloxane composite as superhydrophobic layer were deposited and coated onto cotton fabrics was achieved for water–oil separation application [38], fabrication of superhydrophobic flame retardant coating using depositing method of diammonium phosphate solution, and H-ZrO2 for good wear resistance, acid and alkali resistance and UV resistance excellent and flame-retardant performance [39], fabrication of cotton fabric with fluoroalkylated oligomeric silica/triazine derivative nanocomposites for use in the separation membranes for the separation of oil/water [40], fabrication of superhydrophobic fabric by modifying the pristine fabric with silica nanoparticles prepared from tetraethoxysilane followed by amino-modified polydimethylsiloxane (PDMS) and through a dip-coating process for superhydrophobic and breathable textile applications [41], fabrication of a super-hydrophobic coating on cotton fabric via sol–gel process using super-hydrophobic glycidoxypropyltrimethoxysilane and 1,1,1,3,3,3-hexamethyl disilazane modified TiO2/SiO2 coated cotton fabrics for oil–water separation [42], fabrication of textile with silver chloride nanoparticles under ultrasound irradiation was achieved for antibacterial activity [43].
CF is widely used to produce various industrial products due to its softness, low cost, breathability, and ability to absorb moisture and is a useful starting material for new product development. However, the major drawbacks of cotton are being highly flammable, affected by microbes, and insects, needing chemical modification for wide applications. It is generally more flammable than most of the synthetic fibers. Flame retardants represent one of the most important industrial uses of textile materials and are widely used to make cotton textiles flame resistant for application fields of human safety. One drawback of CF is their flammability with low limited oxygen (LOI = 18.4% only) and their application in firefighters’ protective clothing, military, and airline industry has been restricted [44]. Therefore, the improvement of the thermal stability of the CF (or cotton-based material) becomes one of the important issues.
There are many ways to improve the fire-retardant properties of cotton-based materials such as using flame retardants in polymers or using flame retardant coatings and surface modification. Various flame retardants additives have been applied to modify the combustion characteristics of cotton-based fabrics mainly include inorganic flame retardants such as aluminum hydroxide [45] and magnesium hydroxide [46], nitrogen and phosphorus flame retardants [47, 48].
Recently, the coating onto fabrics using nanocomposite material of flame-retardant properties by the layer-by-layer assembly has attracted attention as a simple and cost-effective process. Cotton and textile materials are properly treated by coating treatments to become waterproof surface and fire-resistant which are useful and necessary for humans in sailing, military endeavors, agricultural greenhouses, and ventures outdoors. The coating processing includes solution immersion, institutional draperies, spray coating, layer-by-layer deposition, sol–gel method, in-situ polymerization, and pad-dry-cure-method [49].
In this work, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites were synthesized via γ-irradiation and used as coating materials to prepare waterproof and flame-retardant CF. The main hypothesis of this study is the conversion of CF from hydrophilic and flammable fabrics to a promising waterproof and flame-retardant ones by utilizing the irradiated EVA and nano SiO2/Al2O3 nanoparticles as coating materials. The physicochemical properties of EVA-based nanocomposites were investigated by estimating the gel content (%) and characterized by Fourier transform infrared (FT-IR), X-ray diffraction (XRD), differential scanning calorimetry (DSC), and Scanning Electron Microscopy (SEM). The effects of as-prepared EVA-based nanocomposites on CF for use in water resistance and fire retardant applications have been studied. The waterproof, flame-retardant properties and thermal stability of treated CF were investigated by mechanical test, thermal gravimetric analysis (TGA), water vapor transmission test, contact angle measurements, and Limiting oxygen index (LOI) and vertical burning flame tests.
2 Experimental
2.1 Materials
Ethylene vinyl acetate copolymer (EVA) containing 10 wt% vinyl acetate, density of 0.935 g/cm3, and melt flow index of 1.3 g/10 min (at 190 °C/2.16 kg) was purchased from Hanhwa Co., South Korea. The silicon dioxide nano-powder (SiO2) with the average particle size of 10–20 nm was purchased from Sigma-Aldrich Co. (China). Aluminum sulfate hexadecahydrate (Al2(SO4)3.16H2O) (98%) (M.W 630.38) was provided by El-Nasr Pharmaceutical Chemicals Company, Egypt. Sodium hydroxide (NaOH), chloroform and toluene were obtained from Qualikems fine chemicals Pvt. Ltd. New Delhi, India. Sample of cotton fabrics (CF) was supplied by Miser Company for Spinning and Weaving, El-Mahala El-Kubra, Egypt.
2.2 Preparation of Al2O3 Nanoparticles
The Al2O3 nanoparticles were prepared by co-precipitation method using aluminum sulfate hexadecahydrate (Al2(SO4)3.16H2O) and sodium hydroxide according to the previously described method [50]. Firstly, 0.1 M of Al2(SO4)3.16H2O was prepared in distilled water using a magnetic stirrer at room temperature. Then, 0.2 M of NaOH was added. A white creamy solution was produced from aluminum hydroxide, separated utilizing a centrifuge. Then, the precipitate was washed several times with distilled water, dried at 80 °C overnight. After that, the dried precipitate was completely converted to Al2O3 nanoparticles by were subsequently annealed at 600 °C for 4 h, then cooled to room temperature and used.
2.3 Preparation of EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 Nanocomposites
EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites were prepared as follows:
Firstly, EVA (10 wt%) was dissolved in chloroform at 70 °C for 30 min with continuous stirring until complete dissolution. Secondly, to prepare EVA/SiO2, 10% SiO2 nanoparticles was dispersed in EVA copolymer solution. To prepare EVA/Al2O3 was prepared by mixing 10% Al2O3 nanoparticles to EVA copolymer solution. EVA/SiO2/Al2O3 nanocomposite was prepared by mixing 5% SiO2 and 5% Al2O3 nanoparticles with EVA copolymer solution. The ratio of SiO2 or Al2O3 nanoparticles was relatively to the concentration of EVA copolymer solution. The EVA mixture solutions with stirred for 3 h at 50 °C until homogeneity, then stirred for another 2 h under sonication to obtain reactant mixture. Thirdly, the reactant mixtures of EVA/10% SiO2, EVA/10% Al2O3 and EVA/5% SiO2/5% Al2O3 as well as pure EVA copolymer were sealed and then exposed to γ-irradiation at different doses of 10, 20, 30, 40, and 50 kGy (dose rate 1.05 kGy/h) using the 60Co Canadian γ-irradiation facility established at the National Center for Radiation Research and Technology, Egyptian Atomic Energy Authority, Cairo, Egypt. Finally, the obtained irradiated mixture solutions (EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3) were casted into Petri-dishes and allowed to dry in air at room temperature for further characterization.
2.4 The Gel Content (%)
The gel content of dried samples of EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites prepared at different irradiation doses of 10–50 kGy were soaked in boiling toluene for 8 h to remove the (uncross-linked) soluble part. Then, the samples were removed, dried to a constant weight, and the insoluble residue was weighed. The gel content (%) was determined according to the following Eq. (1):
where M1 is the sample initial mass and M2 is the residual mass of dried samples after extraction from toluene.
2.5 Treatment of CF
Before processing, CF (15 cm × 15 cm) were washed with tap water to remove some adsorbed impurities then dried at 70 °C for 2 h and the dry weight of CF was measured (W0). After that, the CF were immersed for 30 min in the coating solutions of EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 that prepared by irradiation at 30 kGy. Then the treated CF allowed to dry at 50 °C, furthermore, washed several times with ethanol/distilled water, and dried again at 50 °C. Finally, after deposition and complete evaporation of chloroform (as a solvent), the treated CF weighed (Wg). The weight gain (%) was calculated according to the following Eq. (2):
2.6 Water Absorption (%)
Water absorption was measured on coated CF samples of 5 cm × 5 cm which were dried to constant weight before immersion in distilled water at room temperature for 30 min, then weighted gain with time after absorption. The average water absorption value for three samples was calculated according to the following Eq. (3):
Wd and Ww are the weights of samples dry and wet, respectively.
2.7 Water Vapor Transmission Rate (WVTR)
The WVTR was measured gravimetrically at 40 °C according to the European Pharmacopoeia standard (EP) [51, 52]. WVTR was identified as the amount of water vapor in one gram able to permit through a material, generally within 24 h. Briefly, for this measurement samples of untreated and treated CF were cut into circular pieces with a diameter of 5 cm and put on the mouth of a glass bottle containing 50 mL of distilled water and then the mouth of bottles was secured with Teflon tape. The initial weight was recorded as W1. Then, the bottles were placed in an oven at 40 °C for 24 h. Then the bottles were taken out and weighted (W2). The open bottle served as the negative control. The weight loss (%) of the system reveals an index of WVTR and the WVTR was calculated according to the following Eq. (4):
A (m2) is the area of films covered the area of bottle mouth.
2.8 Characterization Analysis
XRD patterns were obtained with Shimadzu Scientific Instruments (SSI) XRD-6000 series—Kyoto, Japan using Ni-filter and Cu-Kα radiation target (λ = 1.54056 Ao). The pattern was recorded at a scanning rate of 4 steps/min within the range of (2θ) 4° –90°.
The transmittance of the samples was examined utilizing Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy, Bruker Optik GmbH, Ettlingen, Germany), in the range of 4000–400 cm−1.
The size of and morphology of SiO2 and Al2O3 nanoparticles were studied utilizing a Transmission Electron Microscopy (TEM), JEOL JSM–100 CX, Japan, with an acceleration voltage of 80 kV. For TEM investigation, the particle size of SiO2 and Al2O3 samples were set up by making a suspension from the nanoparticles in acetone using an ultrasonic water bath. The suspension was centrifuged to isolate the large size particles. After that, a drop of each suspension (10 µL) was dropped into the carbon-coated copper grid and left to dry at room temperature for further investigatin.
The surface morphology of EVA-based nanocomposites was investigated utilizing Scanning Electron Microscopy (SEM), JEOL JSM-5400, Japan. The surface of the composites was sputter-coated with gold for 3 min.
The tensile strength and elongation at break (%) were measured using dumbbell-shaped pieces of 50 mm long and 4 mm neck width at a crosshead speed of 500 mm/min at room temperature with a tension speed of 25 mm/min, using a tensile testing machine Qchida computerized testing machine, Dongguan Haida Equipment Co. Ltd. China. The ISO 527-2 standards and ASTM D 412a-98 were followed to measure tensile strength and an elongation at break, respectively. The average value of the mechanical parameters was calculated as a mean of three samples.
The thermal analysis of the samples were investigated utilizing the DSC Shimadzu type DSC-50 system in a nitrogen atmosphere at 20 mL/min within the temperature range from ambient to 350 oC at a heating rate of 10 oC/min. Thermogravimetric analysis (TGA) was performed with a Shimadzu TGA-50 framework (Kyoto, Japan) in the temperature range 25−600 °C at a heating rate of 10 oC/min under controlled nitrogen flow of 20 mL/min.
The contact angle between water and treated CF was determined using Biolin Scientific theta lite optical tensiometer by sessile drop test.
The Limiting Oxygen Index (LOI) percentages of the untreated and treated CF were measured using LOI instrument (Rheometric Scientific, UK) according to ASTM D 2863. The analysis was made at the National Institute of Standards, Giza, Egypt. The LOI is the least oxygen (O2) concentration in the oxygen/nitrogen mixture [O2/N2] that either a dimension of 5 cm × 13 cm sample is consumed or flame combustion of the material for 3 min is preserved, with the sample vertically placed (the top of the test sample is inflamed with a burner). The vertical flame tests were conducted on vertical flammability kindle model with strips (1.5 cm × 7.0 cm) of CF and CF treated with EVA and EVA based nanocomposites were subjected directly to the flame at the left bottom according to American Society for Standards and Testing (ASTM D-6413-08) Standard Test Method for Flame Resistance of Textiles (Vertical Flame Test, 2008).
3 Results and Discussion
3.1 Characterization of SiO2 and Al2O3 Nanoparticles
Figure 1A (curve a) shows the XRD of SiO2 with a broad peak at 2θ = 22.7° of amorphous silica [53]. The XRD of the prepared Al2O3 nanoparticles Fig. 1A (curve b shows that Al2O3 appeared together in the form of α- and γ- Al2O3 phases. The α-Al2O3 phase nanoparticles appeared with diffraction peaks at 2θ = 36.1°, 37.6°, 43.9°, 52.5°, 57.8° and 66.2° which are corresponding to the crystal planes of the (104), (110), (113), (024), (116) and (214), respectively. These values correspond exactly to the International Center of Diffraction Data (ICDD) of card number 00–010–0173 of α-Al2O3 phase [54]. Meanwhile, the γ-Al2O3 nanoparticles phase appeared with diffraction peaks at 2θ = 20.2°, 31.2°, 40.5°, 47.4°, 68.7° and 83.5° which are corresponding to the crystal planes of the (111), (220), (222), (400), (440) and (444), respectively. These peaks are in a good agreement with the data of JCPDS cards values corresponding exactly to cubic structure (JCPDS No. 10-0425, 29-0063, and 29-1486) [55]. The average crystallite dimension of Al2O3 nanoparticles from the XRD data of peaks details was determined by applying Debeys–Scherrer Eq. (5)

A XRD, B FT-IR and C TEM characterization of (a) SiO2 and (b) Al2O3 nanoparticles
where D is particle diameter size, k is the so-called shape or geometry factor which equals 0.9, λ is the x-ray wavelength (λ = 0.1541 nm), β is the full width at half maximum (FWHM) of diffraction peak and θ is the diffraction angle. The calculated average crystallite dimension (D) was 31 nm.
Figure 1B (curve a) shows the FT-IR spectrum of the SiO2. The absorption peak at 3424 cm−1 is assigned to the stretching vibration of the hydroxyl group (OH) of the adsorbed water. The peak at 1091 cm−1 is assigned to the Si–O–Si asymmetric stretching vibration. The peak at 814 cm−1 is ascribed to the asymmetric bending and stretching vibration of Si–OH. The peaks at 480 and 652 cm−1 are attributed to the Si–O vibration and O–Si–O bending, respectively [56, 57]. Figure 1B (curve b) shows the FT-IR spectrum of the synthesized Al2O3 nanoparticles. The absorption peak at 3388 cm−1 is due to the stretching mode of bending and stretching hydroxyl groups from the adsorbed water molecules. The peaks at 663–711 cm−1 and 1078 cm−1 are related to Al–O stretching and symmetric bending of Al–O–H, respectively [58,59,60].
Figure 1C presents the TEM micrographs of SiO2 and Al2O3 nanoparticles. The TEM micrograph of SiO2 nanoparticles (Fig. 1C image a) are high dispersion with some aggregation appeared and the particles size in the range of 10–27 nm. Meanwhile, the TEM morphology of Al2O3 nanoparticles (Fig. 1C image b) was irregular spherical shaped, uniformly dispersed and the particles size in the range of 35–63 nm.
3.2 Development and Characterization of EVA Copolymer, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 Nanocomposites
3.2.1 Effect of γ-Irradiation on Gel Content (%)
EVA can be used for various applications but it suffers some disadvantages such as low tensile strength and high flammability. To improve its properties and crosslinking density, it is necessary to modify EVA using additives, fillers, irradiation, or blending with other polymers. The effect of γ-irradiation on either EVA alone or incorporated with conventional nanoparticles such as SiO2 and/or Al2O3 was studied as a function of gel content using hot toluene as a solvent was represented in Fig. 2. Gel content analysis is fundamental to the present research, as it directly measures the crosslinking range of the as-prepared EVA composites. It was noticed that the un-irradiated EVA became completely soluble in hot toluene. The gel content percentage of EVA/SiO2, EVA/Al2O3 and EVA/SiO2/Al2O3 became increasingly enhanced than pure EVA without irradiation because, as the SiO2 and Al2O3 nanoparticle restricted the segmental motion of the EVA polymeric chains, thus raising the cross-linked density. In addition, the formation of the covalent bonds besides the weak van der Waals forces between EVA chains and SiO2/Al2O3 nanocomposite occurred because SiO2 and Al2O3 nanoparticles affected the mobility of the polymer chains due to the bonding between SiO2 and Al2O3 nanoparticles and the polymer chains [61, 62]. However, upon irradiation of EVA copolymer, the gel content percentage is increasingly improved with an increasing radiation dose up to 30 kGy, and then the maximum stable ratio was obtained with further increase in the irradiation dose. On other hand, the use of either SiO2 and/ or conjunction with Al2O3 nanoparticles during the irradiation process provides efficient crosslinking density via increasing the gel content (%). At 30 kGy, the gel content percentage of EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 was 69.1, 81.2, 88.9, and 95.3%, respectively. The irradiation dose of 30 kGy was selected as the optimum dose for the highest crosslinking of EVA copolymer. The increase in the crosslinking of EVA based composite is mainly due to the inter-molecular hydrogen bonding formed between the carbonyl group of acetate groups of EVA and OH groups on the surface of both SiO2 and Al2O3 particles. In addition, during irradiation, molecular radicals are formed, as a result of which they merge into the crosslink. With an increase in the absorbed dose, more total radicals are produced, which leads to greater entanglement of the polymeric chains thus raising the interconnected crosslinking efficiency. The gel content percentage of EVA copolymer was enhanced via γ-irradiation and by incorporation of SiO2 and Al2O3 nanoparticles. It has been stated that the addition of nanosilica could enhance the properties of EVA copolymer [17, 63]. As a result, the gel content percentage of the EVA copolymer was developed by γ-irradiation and was enhanced directly by the incorporation of SiO2 and Al2O3 nanoparticles.

Effect of the irradiation dose on the gel content percentage of EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites
3.2.2 XRD of EVA-Based Nanocomposites
Figure 3 shows the XRD spectra of EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites. The neat EVA sample (Fig. 3a) shows a sharp crystalline peak at 2θ = 20.8°, resulting generally from the crystalline region of EVA caused by the inter-chain hydrogen bonding [64, 65]. The XRD spectra of EVA/SiO2 (Fig. 3b) shows a sharp crystalline peak corresponding to EVA at 2θ = 21.3° and a small peak at 2θ = 23.2° due to the diffraction peak of Sio2. Figure 3c shows the XRD spectra of EVA/Al2O3 that includes a combination of the XRD diffraction peak of EVA copolymer at 2θ = 20.6° as well as the characteristic XRD diffraction peaks of Al2O3 nanoparticles at 2θ = 31.5°, 36.7°, 37.9°, 40.7°, 44.2°, 47.4°, 53.3°, 58°, 66.2°, 69°, and 83.6° which are corresponding to the crystal planes of (220), (104), (110), (222), (113), (400), (024), (116), (214), (440) and (444), respectively. On the other hand, the XRD spectra of EVA/SiO2/Al2O3 nanocomposite is represented in Fig. 3d containing the major corresponding diffraction peaks for EVA, SiO2 and Al2O3. The XRD results confirmed that the successful incorporation of both nanoparticles into EVA copolymer.

XRD spectra of a EVA, b EVA/SiO2, c EVA/Al2O3, and d EVA/SiO2/Al2O3 nanocomposites
3.2.3 FTIR Results of EVA-Based Nanocomposites
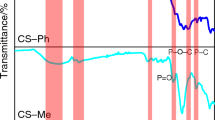
The FTIR spectrum of EVA (Fig. 4a) shows the characteristic peaks attributed to the vinyl acetate groups. The sharp peak at 1735 cm−1 was attributed to the C=O stretching of carbonyl groups. The peak at 1238 cm−1 assigned to C–O–C stretching. The peak at 1021 cm−1 was related to O–C=O out of plane bending of the ester group. The peak at 1371 cm−1 was due to the stretching of the methylene CH2 group. The peak at 1462 cm−1 was due to the bending vibration of the methylene –CH2 group. The peaks at 2850–2917 cm−1 were attributed to the symmetric and asymmetric stretching vibration of methylene [64]. The FTIR results of EVA/SiO2 (Fig. 4b) which demonstrates the change between the spectra of the acetate group of EVA and after addition of SiO2 to EVA. It was noticed that there was a significant shift in C=O stretching of acetate group of EVA in EVA/SiO2 occurred at 1724 cm−1 instead of 1735 cm−1. This could be due to the hydrogen bonding between surface silanol groups (Si–OH) of SiO2 and C=O groups of EVA. In addition, all the characteristic peaks of SiO2 were observed at 1091, 816, 476, and 652 cm−1 due to the absorption peak of Si–O–Si, Si–OH, Si–O, and O–Si–O, respectively.

FTIR spectrum of a EVA, b EVA/SiO2, c EVA/Al2O3 and d EVA/SiO2/Al2O3 nanocomposites
The FTIR spectrum of EVA/Al2O3 (Fig. 4c) represents the same characteristic peaks of EVA in addition to the peak of Al–O stretching that appeared at 713 cm−1 and 1097 cm−1. The interaction between EVA and Al2O3 leads to a significant shift in the C=O stretching of EVA to 1719 cm−1 due to the inter-molecular hydrogen bonding formed between the C=O of acetate groups of EVA and OH groups on the surface of Al2O3 particles. Figure 4d shows the FTIR spectrum of EVA/SiO2/Al2O3 nanocomposite, the C=O stretching of EVA appeared at 1706 cm−1 and this shift was attributed to the incorporation of SiO2 and Al2O3 and inter-molecular hydrogen bonding formed. In addition, the peaks at 1099, 811, 713, and 471 cm−1 are corresponding to Si–O–Si, Si–OH, Al–O, and Si–O, respectively. These results confirmed that SiO2 and Al2O3 were fully incorporated with EVA copolymer.
3.2.4 DSC Results
The thermal behavior and the variations in melting temperature (Tm) of unirradiated EVA, irradiated EVA and EVA/SiO2, EVA/Al2O3 and EVA/SiO2/Al2O3 nanocomposites were investigated using DSC measurements as presented in Fig. 5. The pristine EVA (unirradiated) exhibits a glass transition (Tg) at 50.5 °C, and a melting temperature (Tm) at about 69.4 °C. Meanwhile, upon irradiation of EVA, a shift to higher onset crystallization temperature was achieved. The Tg and Tm increased to 59.3 °C and 76.6 °C, respectively. The irradiation of EVA copolymer enhanced its crystallization and Tm. On the other hand, the DSC curve of EVA/SiO2 showed an increase in the Tm to 172.4 °C upon addition of SiO2 to EVA. This indicates that the addition of small and rigid silica particles could induce enhancement in crystallization of the EVA copolymer. In addition, there was an increase in Tm EVA/Al2O3 to 195.6 oC with the addition of Al2O3 nanoparticles to EVA. However, the Tm of EVA/SiO2/Al2O3 nanocomposite was 187.7 oC in between the Tm of EVA/SiO2 and EVA/Al2O3 nanocomposites.

DSC curves of unirradiated EVA, irradiated EVA, irradiated EVA/SiO2, irradiated EVA/Al2O3, and irradiated EVA/SiO2/Al2O3 nanocomposites
3.2.5 Surface Morphology of EVA-Based Nanocomposites
Figure 6a–d shows the SEM images of EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites irradiated at 30 kGy, respectively. Figure 6a presented that the surface of EVA is dense, tabular, and smooth, and no sign of porous structure. While Fig. 6b shows the SEM image of the EVA/SiO2 nanocomposite, the SiO2 particles were dispersed uniformly in the EVA matrix. The SiO2 particles were embedded in the EVA matrix without aggregation and without phase separation between them. Furthermore, Fig. 6c shows the SEM image of EVA/Al2O3 nanocomposite, where the Al2O3 nanoparticles are easily observed in the microscopic image because of their bigger size compared with the SiO2 particles. EVA/Al2O3 nanocomposite image had a rock-shape surface, rough, and revealed some agglomeration. Additionally, the Al2O3 nanoparticles were dispersed in the EVA matrix with white stone-like and irregular shapes. The SEM image of EVA/Al2O3 showed that Al2O3 nanoparticles are more agglomerated than SiO2 nanoparticles in SEM image of EVA/SiO2. However after addition of SiO2 to EVA/Al2O3, the surface became relative homogeneous and good dispersion was observed as well as the dispersion of both nanoparticles was significantly improved as presented in Fig. 6d for SEM image of EVA/SiO2/Al2O3 nanocomposite, The improved morphology of EVA/SiO2/Al2O3 nanocomposite might be due to the trapping of smaller size of SiO2, which reduces the chance of particle–particle interaction of both nanoparticles; as result dispersion is improved [66]. The SEM images reveal that the homogeneity of nanocomposites increased with the increase of dispersion of the SiO2 nanoparticles.

SEM photographs of a EVA, b EVA/SiO2, c EVA/Al2O3, and d EVA/SiO2/Al2O3 nanocomposites prepared at 30 kGy irradiation dose (magnification factor is 1000X)
3.3 Treatment of CF by EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 Irradiated at 30 kGy for Application as Waterproof and Flame Retardant
3.3.1 Characterization of the Treated CF
3.3.1.1 Evaluation of Weight Gain (%)
The weight gain percentage onto CF after treatment by irradiated EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites is presented in Fig. 7. The treatment was performed by double-layer coating of CF and evaporation of the solvent. It was noted that CF/EVA coated sample exhibited 36.8% weight gain percentage. On the other hand, the treatment of CF with EVA/SiO2 or EVA/Al2O3 or EVA/SiO2/Al2O3 exhibited significantly higher weight gain percentage rather than CF treated with EVA alone. The weight gain percentage of CF samples treated by EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 were 47.2, 58.4, and 53.8%, respectively. The increase in weight gain percentage on CF by EVA based nanocomposites is mainly due to the strong hydrogen bonding between the acetate groups of EVA and OH groups of the surface of silica and alumina particles. Additionally, SiO2 consists of a significant amount of surface silanol groups (Si–OH) which play an important role in forming interaction with polar groups of CF [67].

The weight gain percentage obtained after treatment of CF by irradiated EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3
3.3.1.2 Mechanical Properties of Treated CF
Figure 8 presents the change in the tensile strength and elongation at break percentage of the untreated CF and CF treated with EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites. Compared with control the CF, it was found that treatment of the CF with EVA based nanocomposites enhanced the mechanical properties. The tensile strength of CF with EVA based nanocomposites was higher than both of pure CF and CF treated with EVA alone (CF/EVA). It was found that the tensile strength of pure CF was 10 MPa and the tensile strength of CF/EVA, CF/EVA/SiO2, CF/EVA/Al2O3, and CF/EVA/SiO2/Al2O3 was improved to 17.9, 19.9, 25.4, and 26.5 MPa, respectively. The increase in the tensile strength of treated CF by EVA based nanocomposites as compared to CF and CF/EVA was enhanced via an ascending trend by the presence and interfacial interaction of SiO2 and/or Al2O3 nanoparticles within the EVA matrix. SiO2 is commonly utilized as an improving agent in thermoplastic polymers to improve the mechanical properties, especially the tensile strength [68, 69]. Moreover, Al2O3 nanoparticles and EVA matrix interactions minimize the free movement of CF segments and thus enhance the increase in the tensile strength. This shows that introducing the SiO2 and Al2O3 with EVA was effective and leads to the formation of interactive matrix structures. In contrast, the elongation at break percentage of the treated CF decreased due to the reduction of interweaved interspaces between CF derived from the coating by EVA or EVA based nanocomposites films formed on the surface of fibers. These results produce a wide potential application in the industry of coatings of CF.

Mechanical properties (tensile strength and elongation at break %) of the treated CF samples
3.3.1.3 Thermal Properties of Treated CF
The thermal stability of CF/EVA, CF/EVA/SiO2, CF/EVA/Al2O3, and CF/EVA/SiO2/Al2O3 was investigated by TGA as shown in Fig. 9. The treated CF with EVA (CF/EVA) decomposed in three stages of weight loss percentages, where the first stage is up to 179 °C with weight loss of 5.3% is due to the elimination of water vaporization and elimination of volatile products, the second decomposition stage starts from 180 to 350 °C with weight loss of 30% is attributed to the loss of acetic acid due to the decomposition of vinyl acetate groups, and the third stage starts from between 351 °C and 465 °C with complete decomposition and continuous losing weight which corresponds to the degradation of the polyethylene chains and thermal decomposition of crosslinked backbone and leads to the production of combustible gases and a small amount of char that is finally degraded at higher temperatures. The thermal degradation curves of treated fabrics with CF/EVA/SiO2, CF/EVA/Al2O3 start decomposition in the first stage up to 234 °C and 246 °C with a weight loss of 4.8 and 3.3% respectively. the second decomposition stage starts from 369 °C with a weight loss of 29.6% and the third stage starts from 408 °C and ends at 497 °C leaving 2.6 and 13.8% as a residue of SiO2 and Al2O3, respectively. Meanwhile, the thermal degradation curve of CF/EVA/SiO2/Al2O3 was stable up to 252 °C without any weight loss then start their first stage from 252 to 364 °C with a weight loss of 24%, the second decomposition stage starts from 418 °C leaving 15.7% as a residue of SiO2/Al2O3. Therefore, the TGA results illustrated that the treatment of CF by EVA based nanocomposites, (CF/EVA/SiO2, CF/EVA/Al2O3, and CF/EVA/SiO2/Al2O3), able to advance the thermal stability and had a significant impact on flammability of CF.

TGA curves of CF treated by EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites
3.3.2 Waterproof Application of Treated CF
3.3.2.1 Water Absorption of Treated CF
The hydrophobic surface property of CF was developed by modification utilizing EVA based nanocomposites. The water absorption percentage for control and treated CF was measured as a function of the time up to 30 min as presented in Fig. 10A. It was found that the treated CF had a lower water absorbency. The capability of a material to absorb water is an indirect measure of its hydrophilicity. The water absorption (%) of control CF increases rapidly with the first 5 min., then increases gradually with time until reaches the maximum value after 15 min of 202.4%. However, the coating of CF with EVA lowers the water absorption (%) to 32.6% after 15 min. EVA played an important role in reducing the water absorption of CF about 6.3 folds. In addition the treatment of CF with EVA based nanocomposites played a crucial role in reducing the water absorption percentage of CF to a negligible value of 6–8% after 15 min. within 25 folds in reduction of their ability for water absorptivity. Therefore, the surfaces of the CF were converted from hydrophilic to hydrophobic.

A water absorption behavior of CF and treated CF by γ-irradiated (B) optical image of (a) control CF, (b) CF/EVA, (c) CF/EVA/SiO2, (d) CF/EVA/Al2O3 and (e) CF/EVA/SiO2/Al2O3
The surface morphologies of the CF coated by EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 at low magnification (200X), are presented in Fig. 10B. The surface of pristine CF (Fig. 10B image a) appeared in characteristic parallel texture fibers. After treatment of CF, a thin layer of EVA has fully covered the spaces between texture fibers, even after it was coated with a smooth texture surface without aggregation as presented in Fig. 10B (images b). On the other hand, a dense and stable layer of film is formed on the surface of CF after the modified treatment by EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 as illustrated in Fig. 10B images c-e. Moreover, the CF surface became fully covered compared with the control CF. The fibers in the treated CF held together more tightly and did not easily fall off and separate, because of the crosslinking between fibers and EVA, SiO2 and Al2O3 compounds and a dense layer of film blocked the interstices between fibers. The stable building layer adhered onto the surface of CF by EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 formed over CF may be due to (1) strong van der Waals forces existing between a between EVA, SiO2, Al2O3 and textile fiber of the CF, (2) the formation of waterproof surface and (3) flexible EVA allow conformal adhered to the surface of textile fibers, which maximized the surface contact area between SiO2 and Al2O3 and CF.
3.3.2.2 Contact Angle
The surface wettability of treated CF was examined by contact angle measurements as depicted in Fig. 11. It was noticed that the modification of CF by EVA-based nanocomposites has an important impact on the resulting contact angle of treated CF. It is well known that the contact angle for pristine CF is zero degree because the pristine CF were easily wetted by water drops and spread instantly. Also, this is due to the capillary effect and the abundant hydrophilic hydroxyl groups of CF, that make fabric superhydrophilic [70]. On the other hand, the treatment of CF by EVA (CF/EVA) raised the contact angle to 65° and the net contact angle of EVA is 63.3° [71] which is closest to our results. EVA exhibits superhydrophobic behavior and has a less pronounced hydrophilic nature; therefore, coating of CF with EVA leads to an increase in the contact angle value. Upon treatment of CF by CF/EVA/SiO2, the contact angle was increased to 93° indicating that the addition of SiO2 beside EVA produces a strong water resistance and increased the surface roughness at which the hydrophobicity of treated CF increased. Also, the increase in contact angles of CF/EVA/SiO2 than CF/EVA is due to the increase in surface polarity and this is because the SiO2 surface fully consists of various types of silanol groups (Si–OH) [72]. Moreover, the contact angles of the CF/EVA/Al2O3 and CF/EVA/SiO2/Al2O3 are increased to 98° and 107° indicating hydrophobic surfaces. The increase in the contact angles for all treated CF indicates their affinity towards water decreases and reveals an increase in their hydrophobicity. The results showed that the coated CF has a strong hydrophobic ability. In similar results, it was found that the silk fabric treated with silica composite coatings possessed a good hydrophobic property with an increase in the contact angles [73].

Contact angles of untreated CF and treated CF
3.3.2.3 Water Vapor Permeability
The WVTR was measured gravimetrically and was expressed as an amount of water vapor in gram able to pass through material within 24 h. Air permeability is an important factor in determining the performance of a textile, and therefore, the water vapor permeability of treated CF were determined by means of WVTR. The measured WVPR values of untreated and treated CF compared to open bottle (negative control) as a function of time up to 24 h are plotted in Fig. 12. Generally, the rate at which water vapor passes through CF depends on the nature of the fiber. With hydrophobic fibers, the rate is very slow, whereas, with hydrophilic fibers the rate is relatively fast. After 9 h the WVTR for negative control was 196 g/m2 and initially falls by pure CF to 128 g/m2. The treatment of CF provides a linear decrease in the WVTR. After 9 h time, the WVTR of CF/EVA, CF/EVA/SiO2, CF/EVA/Al2O3, and CF/EVA/SiO2/Al2O3 was 75, 64, 60, and 38 g/m2, respectively. Finally after 24 h, the WVTR became 154. 97, 78, and 55 g/m2, and the pristine CF became 356 g/m2. Analyzing the results of WVTR as shown in Fig. 12 (inset), after 24 h the decrease in WVTR (%) of CF/EVA, CF/EVA/SiO2, CF/EVA/Al2O3, and CF/EVA/SiO2/Al2O3 was 56.7, 72.8, 77.9, and 84.4%, respectively and it is therefore considered particularly significant. The pristine CF displays higher WVTR ability owing to a large number of hydrophilic hydroxyl groups on the cellulose molecules which can form strong hydrogen bonds with the water molecules. After CF was subjected to weight gain by EVA-based nanocomposites, the thickness of the surface increased and the pore size decreased. Therefore, a weak attraction between water molecules and the surface of the treated CF leads to changing the hydrophilic surface of the CF to hydrophobic nature with waterproofing properties. This result is in consistence with previous studies, where the WVTR value decreases after the CF was treated with waterproofing finish [74].

water vapor transmission rates (WVTR) of CF (control) and treated CF
3.3.3 Flame Retardant Application of Treated CF
The flame-retardant properties of the pure CF and treated CF were investigated by the limiting oxygen index (LOI) test and by vertical burning flame as presented in Figs. 13 and 14, respectively. The LOI is a widely utilized method as a simple and precise method to evaluate the flame retardant properties of materials. The LOI values denote the minimum level of oxygen needed to sustain a candle-like flame when a sample is burned in an atmosphere of oxygen and nitrogen. LOI is an indicator of the flammability of material under small flame conditions. The higher the LOI of the material indicate lower tendency to burn. Textiles are considered to be flammable when LOI values are below 21% oxygen in nitrogen and are considered to be flame-retardant when LOI values fall in the range of 26–28%. At these LOI values, flame retarded test fabric samples are expected to pass open flame tests in either the horizontal or vertical direction [75].

LOI percentage of combustion of CF, CF/EVA, CF/EVA/SiO2, CF/EVA/Al2O3, and CF/EVA/SiO2/Al2O3

Photographs of vertical burning flame test for pure CF and treated CF samples after burning time for 25 s
Figure 13 presents the LOI percentages of pure CF and treated CF samples. The LOI value of the pure CF are between 17 and 19% [76]. In this study, the untreated CF and CF treated with EVA (CF/EVA) showed similar LOI values of 18.8% and 19% respectively indicating that the CF and CF/EVA ignited easily in air. It was reported that the LOI value of EVA was 19.2% [17]. It can be seen that the LOI values of treated CF/EVA increased rapidly by the addition of SiO2 and/or Al2O3 from 19 to 29.3% and suggesting a synergistic effect and developing the flame resistance properties which is helpful to the LOI test. The LOI value of CF/EVA/SiO2, CF/EVA/Al2O3, and CF/EVA/SiO2/Al2O3 was increased to 25.3%, 27.5%, and 29.3%, respectively. The results confirmed that treated CF samples afford flame resistance properties especially CF/EVA/SiO2/Al2O3. In a similar results, SiO2 improved the LOI and enhanced the flame retardant of polypropylene [77] and EVA/magnesium hydroxide blends [78].
The flame retardant of the coated CF was examined using a vertical burning flame test, and the photographs of the different samples upon exposure to flame at different times up to 25 s are shown in Fig. 14. The pure CF was ignited immediately after 5 s, burned rapidly after 10 s, and emitted a lot of flame and smoke after 15 s. The same results were observed for CF/EVA; it was ignited and burned rapidly after 15 s without flame and smoke. On the other hand, the treated CF samples by EVA-based nanocomposites showed satisfying results providing a protective and coating layer on the surface of CF, keeping them from being destroyed by further burning. SiO2 and Al2O3 are usually considered to be inert additives in flame-retardant systems by prolonging the burning time. The samples of CF/EVA/SiO2 and CF/EVA/Al2O3 were not ignited until 15 s. Furthermore, the burning time was prolonged and only about 10% of the CF samples ignited after 25 s with the addition of SiO2 or Al2O3. The CF/EVA/SiO2/Al2O3 sample provides the best result even after 25 s there is no ignition. The utilization of SiO2 in conjunction with Al2O3 as flame retardant systems provide an efficient charred layer in resisting a firing in EVA during burning. The modification of the CF with EVA/SiO2/Al2O3 provides a synergistic retardant and contributes to decreasing the flammability and reduces the burning rate at the same time. Jiao and Chen (2011) indicated that the flame-retardant performances of the EVA composites were enhanced by SiO2 [16]. From the results of the LOI and vertical flammability test, it was confirmed that the effectiveness and the stability of CF/EVA/SiO2/Al2O3 against the effect of the flame even after 25 s of exposure and thus it could be used as flame retardant fabrics.
4 Conclusion
In this study, successful modification of cotton fabrics with EVA-based nanocomposites by coating method was achieved to improve the waterproof and fire retardant applications. The coating solution of EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3, was synthesized by γ-irradiation. The thermal and physicochemical characteristic of EVA copolymer was developed by SiO2, Al2O3 nanoparticles and γ-irradiation. An effective coating of the CF by EVA, EVA /SiO2, EVA/Al2O3, and EVA /SiO2/Al2O3 was carried out. The weight gain percentages of the CF after treatment with EVA, EVA/SiO2, EVA/Al2O3, and EVA/SiO2/Al2O3 nanocomposites were 36.8, 47.2, 58.4 and 53.8%, respectively. After treatment of the CF, enhanced properties were attained, namely, the surface became uniform, smooth and the spaces between fabrics were fully covered without aggregation. This is in addition to improved hydrophobic, thermal and mechanical properties. The flame retardant of the treated CF was tested using limited oxygen index (LOI) and the vertical burning flame tests. The LOI values of the CF, CF/EVA, CF/EVA/SiO2, CF/EVA/Al2O3 and CF/EVA/SiO2/Al2O3 were increased to 18.8, 19.0, 25.3, 27.5, and 29.3%, respectively. The novel aspect and hypothesis of the present study was confirmed by the conversion of the CF from hydrophilic and flammable fabrics to hydrophobic and flame resistant via surface coating with EVA/SiO2/Al2O3 nanocomposite prepared by γ-irradiation. The enhancement in the thermal, mechanical properties, LOI, permeability, contact angle, and water vapor characteristics of CF gives a promising aspect for the utilizing of the CF coated with EVA/SiO2/Al2O3 as flame retardant and water-resistant fabrics for wide potential applications of cotton fabrics.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Y. Bai, J. Qian, Q. An, Z. Zhu, P. Zhang, Pervaporation characteristics of ethylene–vinyl acetate copolymer membranes with different composition for recovery of ethyl acetate from aqueous solution. J. Membr. Sci. 305(1–2), 152–159 (2007)
A.V. Kostyuk, N.M. Smirnova, S.V. Antonov, S.O. Ilyin, Rheological and Adhesion Properties of Hot-Melt Adhesives Based on Hydrocarbon Resins and Poly (ethylene-vinyl acetate). Polym. Sci., Ser. A 63(3), 283–295 (2021)
H. Vahabi, F. Gholami, V. Karaseva, F. Laoutid, R. Mangin, R. Sonnier, M.R. Saeb, Novel nanocomposites based on poly (ethylene-co-vinyl acetate) for coating applications: the complementary actions of hydroxyapatite, MWCNTs and ammonium polyphosphate on flame retardancy. Prog. Org. Coat. 113, 207–217 (2017)
M.A. Bahattab, J. Mosnáček, A.A. Basfar, T.M. Shukri, Cross-linked poly (ethylene vinyl acetate)(EVA)/low density polyethylene (LDPE)/metal hydroxides composites for wire and cable applications. Polym. Bull. 64(6), 569–580 (2010)
D. Lopes, M.J. Ferreira, R. Russo, J.M. Dias, Natural and synthetic rubber/waste–Ethylene-Vinyl Acetate composites for sustainable application in the footwear industry. J. Clean. Prod. 92, 230–236 (2015)
C. Schneider, R. Langer, D. Loveday, D. Hair, Applications of ethylene vinyl acetate copolymers (EVA) in drug delivery systems. J. Control. Release 262, 284–295 (2017)
M. Amini, A.R. Sa, S.A. Haddadi, A. Kheradmand, Mechanical, rheological and oxygen barrier properties of ethylene vinyl acetate/diamond nanocomposites for packaging applications. Diam. Relat. Mater. 99, 107523 (2019)
Z. Li, X. Qi, L. Xu, H. Lu, W. Wang, X. Jin, Z.I. Md, Y. Zhu, Y. Fu, Q. Ni, Self-repairing, large linear working range shape memory carbon nanotubes/ethylene vinyl acetate fiber strain sensor for human movement monitoring. ACS Appl. Mater. Interfaces. 12(37), 42179–42192 (2020)
V. Chaudhary, S. Sharma, Suspension polymerization technique: parameters affecting polymer properties and application in oxidation reactions. J. Polym. Res. 26(5), 1–12 (2019)
X. Dai, L. Yu, Y. Zhang, L. Zhang, J. Tan, Polymerization-induced self-assembly via RAFT-mediated emulsion polymerization of methacrylic monomers. Macromolecules 52(19), 7468–7476 (2019)
D. Horák, N. Semenyuk, F. Lednický, Effect of the reaction parameters on the particle size in the dispersion polymerization of 2-hydroxyethyl and glycidyl methacrylate in the presence of a ferrofluid. J. Polym. Sci., Part A: Polym. Chem. 41(12), 1848–1863 (2003)
A.M. Elbarbary, Y.H. Gad, Radiation synthesis and characterization of poly (vinyl alcohol)/acrylamide/TiO2/SiO2 nanocomposite for removal of metal ion and dye from wastewater. J. Inorgan. Organometal. Polymers Mater. 1–23 (2021)
A.M. Elbarbary, I.A. Ibrahim, H.M. Shafik, S.H. Othman, Magnetic 99mTc-core-shell of polyethylene glycol/polyhydroxyethyl methacrylate based on Fe3O4 nanoparticles: Radiation synthesis, characterization and biodistribution study in tumor bearing mice. Adv. Powder Technol. 28(8), 1898–1910 (2017)
Y.H. Gad, A.M. Elbarbary, Radiation synthesis of Fe3O4/SiO2/glycidyl methacrylate/acrylonitrile nanocomposite for adsorption of basic violet 7 dye: Kinetic, isotherm, and thermodynamic study. Appl. Organomet. Chem. 35(7), e6258 (2021)
N.M. El-Sawy, A.M. Elbarbary, Synthesis and characterization of novel trifunctionalized monomer and their polymer by radiation induced polymerization. J. Macromol. Sci. Part A 49(3), 207–213 (2012)
C.M. Jiao, X.L. Chen, Influence of fumed silica on the flame-retardant properties of ethylene vinyl acetate/aluminum hydroxide composites. J. Appl. Polym. Sci. 120(3), 1285–1289 (2011)
Y. Qian, S. Zhou, X. Chen, Synergistic flame retardant effect between nano-silicon dioxide and layered double hydroxides in ethylene vinyl acetate composites. J. Thermoplast. Compos. Mater. 31(10), 1295–1309 (2018)
A. Walong, B. Thongnuanchan, T. Sakai, N. Lopattananon, Influence of silicon dioxide addition and processing methods on structure, thermal stability and flame retardancy of EVA/NR blend nanocomposite foams. Progress Rubber Plastics Recycling Technol. 37(1), 49–65 (2021)
G. Schmaucks, B. Friede, H. Schreiner, J.O. Roszinski, Amorphous silicon dioxide as additive to improve the fire retardancy of polyamides. Fire Retardancy of Polymers New Strategies and Mechanisms 35–48 (2009)
M. Rostami, M. Mohseni, Z. Ranjbar, An attempt to quantitatively predict the interfacial adhesion of differently surface treated nanosilicas in a polyurethane coating matrix using tensile strength and DMTA analysis. Int. J. Adhes. Adhes. 34, 24–31 (2012)
K. Yatsui, T. Yukawa, C. Grigoriu, M. Hirai, W. Jiang, Synthesis of ultrafine γ-Al2O3 powders by pulsed laser ablation. J. Nanopart. Res. 2(1), 75–83 (2000)
S.R. Smith, R. Rafati, A.S. Haddad, A. Cooper, H. Hamidi, Application of aluminium oxide nanoparticles to enhance rheological and filtration properties of water based muds at HPHT conditions. Colloids Surf., A 537, 361–371 (2018)
N. Wei, J. Hu, M. Zhang, J. He, P. Ni, Cross-linked porous polymer separator using vinyl-modified aluminum oxide nanoparticles as cross-linker for lithium-ion batteries. Electrochim. Acta 307, 495–502 (2019)
M.H. Parvin, J. Arjomandi, J.Y. Lee, γ-Al2O3 nanoparticle catalyst mediated polyaniline gold electrode biosensor for vitamin E. Catal. Commun. 110, 59–63 (2018)
A. Gupta, A. Kumar, K.V. Sharma, R. Gupta, Application of high conductive nanoparticles to enhance the thermal and mechanical properties of wood composite. Mater. Today: Proceed. 5(1), 3143–3149 (2018)
A. Laachachi, M. Ferriol, M. Cochez, J.M.L. Cuesta, D. Ruch, A comparison of the role of boehmite (AlOOH) and alumina (Al2O3) in the thermal stability and flammability of poly (methyl methacrylate). Polym. Degrad. Stab. 94(9), 1373–1378 (2009)
Z. Wen, M. Wu, T. Itoh, M. Kubo, Z. Lin, O. Yamamoto, Effects of alumina whisker in (PEO) 8–LiClO4-based composite polymer electrolytes. Solid State Ionics 148(1–2), 185–191 (2002)
S. Ju, M. Chen, H. Zhang, Z. Zhang, Dielectric properties of nanosilica/low-density polyethylene composites: the surface chemistry of nanoparticles and deep traps induced by nanoparticles, Exp. Polymer Lett. 8(9) (2014).
F.I. Abou El Fadl, A.M. Elbarbary, Radiation synthesis and characterization of heterogeneous magnetic nanocomposites of 2-hydroxyethyl methacrylate for catalytic degradation of sandocryl blue dye. Sep. Purif. Technol. 272, 118972 (2021)
M.A. Elhady, A. Abdeldaym, Effect of aluminum oxide nanoparticles additives and gamma irradiation on the structural and optical properties of syndiotactic polystyrene. Polym. Eng. Sci. 59(3), 555–565 (2019)
R. Li, G. Wu, Preparation of polysaccharide-based hydrogels via radiation technique (Elsevier, Hydrogels based on natural polymers, 2020), pp. 119–148
A.G. Chmielewski, M. Haji-Saeid, S. Ahmed, Progress in radiation processing of polymers. Nucl. Instrum. Methods Phys. Res., Sect. B 236(1–4), 44–54 (2005)
Z. Zhang, Z. Ma, Q. Leng, Y. Wang, Eco-friendly flame retardant coating deposited on cotton fabrics from bio-based chitosan, phytic acid and divalent metal ions. Int. J. Biol. Macromol. 140, 303–310 (2019)
B. Edwards, A. El-Shafei, P. Hauser, P. Malshe, Towards flame retardant cotton fabrics by atmospheric pressure plasma-induced graft polymerization: Synthesis and application of novel phosphoramidate monomers. Surf. Coat. Technol. 209, 73–79 (2012)
Z. Fanglong, X. Qun, F. Qianqian, L. Rangtong, L.I. Kejing, Influence of nano-silica on flame resistance behavior of intumescent flame retardant cellulosic textiles: remarkable synergistic effect? Surf. Coat. Technol. 294, 90–94 (2016)
S. Ghosh, S. Ganguly, P. Das, T.K. Das, M. Bose, N.K. Singha, A.K. Das, N.C. Das, Fabrication of reduced graphene oxide/silver nanoparticles decorated conductive cotton fabric for high performing electromagnetic interference shielding and antibacterial application. Fibers Polymers 20(6), 1161–1171 (2019)
S. Ghosh, S. Ganguly, S. Remanan, N.C. Das, Fabrication and investigation of 3D tuned PEG/PEDOT: PSS treated conductive and durable cotton fabric for superior electrical conductivity and flexible electromagnetic interference shielding. Compos. Sci. Technol. 181, 107682 (2019)
W. Guo, X. Wang, J. Huang, Y. Zhou, W. Cai, J. Wang, L. Song, Y. Hu, Construction of durable flame-retardant and robust superhydrophobic coatings on cotton fabrics for water-oil separation application. Chem. Eng. J. 398, 125661 (2020)
H. Qin, X. Li, X. Zhang, Z. Guo, Preparation and performance testing of superhydrophobic flame retardant cotton fabric. New J. Chem. 43(15), 5839–5848 (2019)
K. Yamashita, A. Yasukawa, H. Sawada, Fabrication of cotton fabric with superoleophilic/superhydrophobic characteristic on the modified surface by using fluoroalkylated oligomeric silica/triazine derivative nanocomposites. Coatings 10(2), 174 (2020)
M. Hasanzadeh, H. Shahriyari Far, A. Haji, G. Rosace, Surface modification of polyester/viscose fabric with silica hydrosol and amino-functionalized polydimethylsiloxane for the preparation of a fluorine-free superhydrophobic and breathable textile. Coatings 12(3), 398 (2022)
P.K. Jaseela, K.O. Shamsheera, A. Joseph, HMDS–GPTMS modified titania silica nanocomposite: a new material for oil–water separation. J. Inorg. Organomet. Polym Mater. 30(6), 2134–2141 (2020)
A.R. Abbasi, M. Bohloulzadeh, A. Morsali, Preparation of AgCl nanoparticles@ ancient textile with antibacterial activity under ultrasound irradiation. J. Inorg. Organomet. Polym Mater. 21(3), 504–510 (2011)
Z. Shariatinia, N. Javeri, S. Shekarriz, Flame retardant cotton fibers produced using novel synthesized halogen-free phosphoramide nanoparticles. Carbohyd. Polym. 118, 183–198 (2015)
M. Goudarzi, D. Ghanbari, N.M. Salavati, Room temperature preparation of aluminum hydroxide nanoparticles and flame retardant poly vinyl alcohol nanocomposite (2015)
Y. Yang, M. Niu, J. Li, B. Xue, J. Dai, Preparation of carbon microspheres coated magnesium hydroxide and its application in polyethylene terephthalate as flame retardant. Polym. Degrad. Stab. 134, 1–9 (2016)
W. Xu, G. Wang, X. Zheng, Research on highly flame-retardant rigid PU foams by combination of nanostructured additives and phosphorus flame retardants. Polym. Degrad. Stab. 111, 142–150 (2015)
N. Wen, W. Zeng, Y. Yang, Z. Yang, H. Li, X. Li, Q. Li, H. Ding, Z. Lei, Preparation of the intrinsic flame-retardant curing agent of inorganic epoxy resin containing nitrogen and phosphorus. J. Inorgan. Organometal. Polymers Mater. 1–11 (2022)
M. Zahid, G. Mazzon, A. Athanassiou, I.S. Bayer, Environmentally benign non-wettable textile treatments: a review of recent state-of-the-art. Adv. Coll. Interface. Sci. 270, 216–250 (2019)
D. Manyasree, P. Kiranmayi, R. Kumar, Synthesis, characterization and antibacterial activity of aluminium oxide nanoparticles. Int. J. Pharm. Pharm. Sci 10(1), 32–35 (2018)
M. Kokabi, M. Sirousazar, Z.M. Hassan, PVA–clay nanocomposite hydrogels for wound dressing. Eur. Polymer J. 43(3), 773–781 (2007)
M.T. Khorasani, A. Joorabloo, A. Moghaddam, H. Shamsi, Z. MansooriMoghadam, Incorporation of ZnO nanoparticles into heparinised polyvinyl alcohol/chitosan hydrogels for wound dressing application. Int. J. Biol. Macromol. 114, 1203–1215 (2018)
T.-H. Liou, F.-W. Chang, J.-J. Lo, Pyrolysis kinetics of acid-leached rice husk. Ind. Eng. Chem. Res. 36(3), 568–573 (1997)
A.A. Mohammed, Z.T. Khodair, A.A. Khadom, Preparation and investigation of the structural properties of α-Al2O3 nanoparticles using the sol-gel method. Chem. Data Collect. 29, 100531 (2020)
A.S. Jbara, Z. Othaman, A.A. Ati, M.A. Saeed, Characterization of γ-Al2O3 nanopowders synthesized by Co-precipitation method. Mater. Chem. Phys. 188, 24–29 (2017)
Y.-X. Wei, C. Deng, Z.-Y. Zhao, Y.-Z. Wang, A novel organic-inorganic hybrid SiO2@ DPP for the fire retardance of polycarbonate. Polym. Degrad. Stab. 154, 177–185 (2018)
W. Xu, G. Wang, Y. Liu, R. Chen, W. Li, Zeolitic imidazolate framework-8 was coated with silica and investigated as a flame retardant to improve the flame retardancy and smoke suppression of epoxy resin. RSC Adv. 8(5), 2575–2585 (2018)
S.A. Naayi, A.I. Hassan, E.T. Salim, FTIR and X-ray diffraction analysis of Al2O3nanostructured thin film prepared at low temperature using spray pyrolysis method. Int. J. Nanoelectron. Mater. 11, 1–6 (2018)
N. Avci, P.F. Smet, J. Lauwaert, H. Vrielinck, D. Poelman, Optical and structural properties of aluminium oxide thin films prepared by a non-aqueous sol–gel technique. J. Sol-Gel. Sci. Technol. 59(2), 327–333 (2011)
C.A. Roy, C.G. Takoudis, Investigation of the aluminum oxide/Si (100) interface formed by chemical vapor deposition. Thin Solid Films 446(1), 155–159 (2004)
A.A.P. Mansur, O.L. Do Nascimento, H.S. Mansur, Enhancing polymer-modified mortar adhesion to ceramic tile surface by chemical functionalization with organosilanes. Surf. Rev. Lett. 16(01), 127–139 (2009)
A.A.P. Mansur, O.L.D. Nascimento, W.L. Vasconcelos, H.S. Mansur, Chemical functionalization of ceramic tile surfaces by silane coupling agents: polymer modified mortar adhesion mechanism implications. Mater. Res. 11(3), 293–302 (2008)
D.Q. Tham, V.M. Tuan, D.T.M. Thanh, N.T. Chinh, N.V. Giang, N.T.T. Trang, T.T.X. Hang, H.T. Huong, N.T.K. Dung, T. Hoang, Preparation and properties of ethylene vinyl acetate copolymer/silica nanocomposites in presence of EVA-G-Acrylic Acid. J. Nanosci. Nanotechnol. 15(4), 2777–2784 (2015)
M.A. Zamiri, A. Kargari, H. Sanaeepur, Ethylene vinyl acetate/poly (ethylene glycol) blend membranes for CO2/N2 separation. Greenhouse Gases: Sci. Technol. 5(5), 668–681 (2015)
M. Sadeghi, G. Khanbabaei, A.H.S. Dehaghani, M. Sadeghi, M.A. Aravand, M. Akbarzade, S. Khatti, Gas permeation properties of ethylene vinyl acetate–silica nanocomposite membranes. J. Membr. Sci. 322(2), 423–428 (2008)
M. Khan, A.A. Khurram, T. Li, T. Zhao, T. Subhani, I.H. Gul, Z. Ali, V. Patel, Synergistic effect of organic and inorganic nano fillers on the dielectric and mechanical properties of epoxy composites. J. Mater. Sci. Technol. 34(12), 2424–2430 (2018)
B. Xu, J. Ding, L. Feng, Y. Ding, F. Ge, Z. Cai, Self-cleaning cotton fabrics via combination of photocatalytic TiO2 and superhydrophobic SiO2. Surf. Coat. Technol. 262, 70–76 (2015)
E. Kontou, M. Niaounakis, Thermo-mechanical properties of LLDPE/SiO2 nanocomposites. Polymer 47(4), 1267–1280 (2006)
T. Buddi, B.N. Rao, S.K. Singh, R. Purohit, R.S. Rana, Development and analysis of high density poly ethylene (HDPE) nano SiO2 and wood powder reinforced polymer matrix hybrid nano composites. J. Exp. Nanosci. 13(sup1), S24–S30 (2018)
C.-H. Xue, Q.-Q. Fan, X.-J. Guo, Q.-F. An, S.-T. Jia, Fabrication of superhydrophobic cotton fabrics by grafting of POSS-based polymers on fibers. Appl. Surf. Sci. 465, 241–248 (2019)
S.T. Palakattukunnel, S. Thomas, P.A. Sreekumar, S. Bandyopadhyay, Poly (ethylene-co-vinyl acetate)/calcium phosphate nanocomposites: contact angle, diffusion and gas permeability studies. J. Polym. Res. 18(6), 1277–1285 (2011)
A. Comas-Vives, Amorphous SiO2 surface models: energetics of the dehydroxylation process, strain, ab initio atomistic thermodynamics and IR spectroscopic signatures. Phys. Chem. Chem. Phys. 18(10), 7475–7482 (2016)
S. Zhao, R. He, X. Zhang, W. Zeng, T. Zhang, W. Yu, H. Liu, Fabrication of reinforced hydrophobic coatings for the protection of silk fabric. Text. Res. J. 89(18), 3811–3824 (2019)
M. Palanikkumaran, A.K. Agrawal, M. Jassal, Water-proof Breathable Coatings Based on Poly (vinyl alcohol) for Cellulosic Fabric. J. Ind. Text. 38(2), 151–166 (2008)
D. Purser, Toxicity of fire retardants in relation to life safety and environmental hazards, Fire retardant Mater. 69–127 (2001)
S. Chang, B. Condon, J. Smith, M. Easson, Preparation of flame retardant cotton fabric using environmental friendly supercritical carbon dioxide. Int. J. Mater. Sci. Appl. 6(6), 269 (2017)
P. Wei, J. Hao, J. Du, Z. Han, J. Wang, An investigation on synergism of an intumescent flame retardant based on silica and alumina. J. Fire Sci. 21(1), 17–28 (2003)
M. Fu, B. Qu, Synergistic flame retardant mechanism of fumed silica in ethylene-vinyl acetate/magnesium hydroxide blends. Polym. Degrad. Stab. 85(1), 633–639 (2004)
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). Not applicable.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests and non-financial competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elbarbary, A.M., Elhady, M.A. & Gad, Y.H. Development of Cotton Fabrics via EVA/SiO2/Al2O3 Nanocomposite Prepared by γ-Irradiation for Waterproof and Fire Retardant Applications. J Inorg Organomet Polym 32, 4039–4056 (2022). https://doi.org/10.1007/s10904-022-02395-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-022-02395-w