
Abstract
In this study, Knoevenagel condensation reaction was performed to prepare a new designed silane coupling agent from the reaction of the synthetic 4-H pyran derivative (EA) and 3-isocyanatopropyl triethoxysilane. The surface modification of silica nanoparticles by novel modifier was achieved by sol–gel technology. Also, in the next step, PVA/silica nanocomposites have been fabricated to investigate the chemical and physical properties of the new decorated nanoparticles. The surface modification of silica nanoparticles reduced the aggregation of nanoparticles effectively using newly prepared silane coupling agent and showed the enhanced performance for host matrix. FT-IR analysis demonstrated that the six membered ring moieties have been covalently bonded to the surface of the SiO2 nanoparticles. The PVA/SiO2 nanocomposites including 2, 5, 10 and 15 wt% nano contents have been prepared. Characterization of the prepared samples with FT-IR, field emission scanning electron microscopy (FE-SEM), and atomic force microscopy (AFM) confirmed the synthesis of the nanocomposites with good dispersion properties for nanoparticles. The particles size of ornamented nanoparticles in nanocomposites are about 50–60 nm as characterized by FE-SEM and AFM analyses.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Multicomponent reactions have been developed as a powerful tool for the synthesis of structurally interesting materials for applications in nanotechnology [1]. Among the different kind of materials, 4H-pyrans are an important category of heterocycles due to many pharmacological, anti-inflammatory and biochemical properties. Therefore, the synthesis of 4H-pyrans and their derivatives have attracted much attention in recent years [2,3,4,5]. For example, Aytemir et al. [6] prepared a series of novel 3-hydroxy-6-methyl-2-substituted 4H-pyran-4-one derivatives as anticonvulsant agents. The anticonvulsant actions of prepared samples were determined in vivo by maximal electroshock (MES), subcutaneous Metrazol (scMet), and rotorod toxicity experiments for neurological deficits. Compound 2-[4-(4-Chloro-phenyl)-piperazin-1-ylmethyl]-3-hydroxy-6-methyl-4H-pyran-4-one was found to be protective against MES and could be valuable in several medicinal applications. Also the antifungal activity was reported by Aytemir group for some of these derivatives. Saundane et al. [7] was synthesized the 2-amino-4-(5′-substituted 2′-phenyl-1H-indol-3′-yl)-6-aryl-4H-pyran-3-carbonitrile derivatives, and presented them as antimicrobial and antioxidant agents. Due to its bi-functionality nature, silane coupling agents are often used to modify the different oxide nanoparticle surfaces [8]. These coupling agents are able to link chemically to the hydroxyl units of oxide surfaces by hydrolysis reaction and preserve the defined spaces between the nano-materials in shape and prevent the agglomeration of these structures in the real applications media [9]. In fact the silicone modifiers provided separated zone between oxide structure and the other structures throughout the organic or organic–inorganic segments by interaction with the spacer structure [10]. Actually the newly created functional groups on the surfaces of the nanoparticles can possibly guarantee the maximum compatibility between organic modified oxide particles and organic host matrices through the better interaction with functional units with matrices linkages. The literature survey procedure revealed that the silicon containing commercially available compounds as silane coupling agents could provide the different organo functional groups including thiol, amine, epoxide, acrylic and etc. on the surface of nanoparticles by condensation reaction [11,12,13]. But the diversities of these structures with defined tasks were limited. Also the creation of new segments bearing specific molecules on the surface of metal oxide nanoparticles has been rarely reported in the literature. The 4-H pyran derivative grafted to the surface of silica nanoparticles as a new silane modifier reduced the aggregation of the nanoparticles by the steric repulsion forces and improved the dispersion properties of nanoparticles through interaction enhancement with the matrix. Also the owning properties of 4-H pyran molecules were shielded and kept for final application area in complexes. In recent years, many researchers studied the combinational properties of dispersed nano-materials in polymeric matrices as nanocomposites [14]. The prepared constructions expanded the bonus of both organic systems as the flexible matrices and inorganic moieties as dispersants. Numerous polymeric matrices have been inspected as the organic phase of the hybrids [15,16,17,18]. Polyvinyl alcohol (PVA) is a popular polymer with biological friendly characteristics and has been developed for bio-based applications such as synthetic wound dressing, artificial skin, and cardiovascular device [19]. PVA is an innocuous, optically transparent, water soluble, biocompatible and environmental friendly polymer with excellent film forming properties. Furthermore vinyl alcohol is a valuable building block due to the reducing ability of the secondary alcohol groups and it has been recognized as a suitable host polymer matrix for nanoparticles [19,20,21,22]. Numerous oxides of transition elements like copper, zinc, titanium, etc. have been used to formulate the nanocomposites using PVA as a known matrix and their syntheses, characterizations, thermal decomposition behaviors and mechanical properties have been extensively studied and stated in the literature [23,24,25,26]. Our investigation in nanoparticles functionalization and characterization directed to the novel synthesis of trialkoxysilane compound as surface modifier for the treatment of metal oxide silica nanoparticles to produce organic modified silica. Based on Asghari et al. experience in the preparations of organic molecules with special features for antibacterial and pharmaceutical applications [27], the novel modifier contains active moiety was prepared for treatment on silica nanoparticles to attain the biologically active nanocomposites with carrier properties. In this work, synthesis and characterization of the organic 4-H pyran structure was achieved in the first step. The new structure Ethyl 4-(2-chlorophenyl)-6-(hydroxymethyl)-8-oxo-2-(3-(3-(triethoxysilyl)propyl)ureido)-4,8-dihydropyrano[3,2-b]pyran-3-carboxylate (UPC) was prepared from the reaction of ethyl 2-amino-4-(2-chlorophenyl)-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carboxylate (EA) and Triethoxy(3-isocyanatopropyl)silane (ICTES). Subsequently, the novel silane modifier was applied to partially bond to the surface of silica nanoparticles. Finally, fabrication of the PVA/silica nanocomposites and the effects of the novel silicon modifier on the PVA nanostructure properties have been investigated. The effects of surface treatment process on the morphological, thermal and physical properties of the nanocomposites were analyzed using FT-IR, UV–Vis, FE-SEM, AFM and TGA. Also AFM analysis was used to investigate the effect of the prepared 4-H pyran derivative-SiO2 dopant on the morphology of the PVA/SiO2 bionanocomposites.
2 Experimental
2.1 Materials
All chemicals were purchased from Merck, Fluka, and Aldrich chemical companies. The PVA powder (Mw = 75,000 Da and degree of hydrolysis, DH = 98%) was obtained from Merck Chemical Co. and was used as received. The 3-isocyanatopropyl triethoxysilane (ICTES) obtained from Fluka Chemical Co. and used as obtained without further purification. Tetrahydrofuran (THF) was purified by refluxing and distilling from calcium hydride. All other reagents, of analytical-grade quality, were commercial products and used as received.
2.2 Characterization
A Jasco-680 Fourier transform infrared (FT-IR) spectroscope (Japan) was employed to examine the chemical bonds on the nanopartcles, surface modified nanoparticles, polymer and nanocomposites. Solids spectra were obtained with KBr pellets. Vibration bands were reported as wavenumber (cm−1). The dispersion morphology of the nanoparticles on PVA matrix was observed using field emission scanning electron microscopy [FE-SEM, HITACHI (S-4160)]. UV–Vis spectrophotometer (Cecil 5000 UV) was employed to gain the absorbance and transmittance behavior of the organic molecules, polymer and nanocomposite films in the wavelength range of 200–500 nm and the scan rate of 5 nm/min. The films for UV–Vis absorbance measurements were organized by dividing films into pieces with a knife and were dried by vacuum at 50 °C for 10 h. The AFM topographic pictures were obtained using digital multimode instruments Compact Frame, Bruker.
2.3 Synthesis of Organic Linker (EA)
The organic linker, namely Ethyl 2-amino-4-(2-chlorophenyl)-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carboxylate (EA) has been synthesized according to the procedure reported in our pervious article [27]. Concisely, 1 mmol of Kojic acid dissolved in water, 1 mmol of ethyl cyanoacetate, 1 mmol of 2-chloro benzaldehyde and 20 mol% of potassium carbonate were added in a two necked round-bottomed flask. The reaction mixture was stirred for 2 h at room temperature. The reaction progress was monitored by thin-layer chromatography and the prepared product was filtered and recrystallized by hexane/ethyl acetate (9:1). The yield of the reaction was over 97%.
2.4 Production of Novel Silane Coupling Agent (UPC)
5 mmol of EA, 5 mmol of ICTES, 10 mL of tetrahydrofuran and three drops of triethylamine as catalysts were mixed in a three necked round-bottomed flask equipped with a mechanical stirrer, a nitrogen inlet and reflux condenser. The mixture was stirred and refluxed for 7 h. The solution precipitated in hexane, and UPC was dried under vacuum at 30 °C for 24 h. The reaction yield was over 95%.
2.5 Reaction of Surface Modifier (UPC) with Silica Nanoparticles
The process of surface modification has been described in our previous study [22]. In that study the commercial silica nanoparticles were modified by synthetic benzothiazole derived silane coupling agent. Here, in-situ preparation of silica nanoparticles was targeted in which SiO2 nanoparticles were prepared by acid catalyzed reaction using tetra-ethyl orthosilicate (TEOS) in ethanol solution [28]. Briefly approximately 30 mL of TEOS is poured into a 100 mL flask and exactly 31 mL of Ethanol were added. The solution was mixed by magnetic stirrer. Then 38 mL of distilled water and 3–4 drop of concentrated HCl is added to the TEOS solution. The solution was stirred for 1.5 h at 60 °C. The molar ratio of TEOS:Ethanol:Water is 1:4:16. Subsequently the certain amount of UPC was added to the mixture and sonicated for 20 min. The mixture was separated, washed with ethanol three times and stored in the dark cool situation. According to the dynamic light scattering (DLS) analysis result the z-average size of the nanoparticles was 53.6 nm.
2.6 Construction of the Modified SiO2/PVA Nanocomposites
5, 10 and 15 wt% of the organic modified nanoparticles were mixed with PVA solution. SiO2/PVA blends were stirred overnight and then casted. The resultant samples were spread in suitable cast. The samples were kept back at room temperature until dried. The fabricated nanocomposites were separated and dried at 70 °C under vacuum for 2 h.
3 Results and Discussion
3.1 Synthesis of Novel Silica Modifier
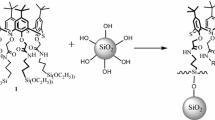
The preparation method of 4-H pyran derivative, EA, is shown in Scheme 1. EA structure was prepared by multicomponent reaction of three different compounds in green media as described in Scheme 1. The reaction is known as Knoevenagel condensation reaction. The spectroscopic data related to the success in the synthesis of EA was reported in previous study [27]. After that, EA was reacted with ICTES to form the UPC structure. The UPC molecule contains different functional units such as amine, hydroxyl and others. Among the different mentioned units in the structure of EA, the amine group showed better tendency to react with isocyanate unit of ICTES. Therefore, the amine functional group in the 4-H pyran component was reacted with isocyanate unit of ICTES to form urea linkage. The prepared structure contains a urea functional unit and was introduced as an active species of 4-H pyran silane coupling agent and used to modify the surface of silica nanostructures.

Schematic illustration of the preparation of the EA, novel silane modifier as UPC, and surface modification of SiO2 nanoparticles (OMSN)
FT-IR spectroscopy was used to distinguish the nature of linkages between ICTES and EA structure. The identification analyses of EA structure were reported in our pervious article [27]. FT-IR spectrum of UPCshowed two broad signals at 3386 and 3327 cm−1 corresponding to amine and hydroxyl functional units, respectively. The FT-IR spectrum obviously revealed bands at 1735, 1674 and 1633 cm−1 as reported in our earlier study, which were generally attributed to the carbonyl stretching of ester, ketone and urea groups, respectively. Also the related peaks of O–Si, and C–Cl bonds stretching vibrations were detected at 1106 and 758 cm−1. These vibrations showed the reaction between ICTES and pendent 4-H pyran. 1H-NMR and 13C-NMR data of the UPCare reported in Table 1. Three signals in 1H-NMR at 3.19, 1.65 and 0.64 ppm which related to the alpha, beta and gamma methylene groups of urea unit respectively were clearly detected. The methylene and methyl substituent protons of triethoxy silane appeared as quartet and triplet at 3.83 and 1.24 ppm, respectively. A triplet signal at 1.04 ppm for the methyl group, a quartet at 3.99 ppm for the methylene group of the ethoxy moiety, a singlet at 4.82 ppm for the CH2 group of CH2OH, a triplet at 5.21 ppm for the OH group, a singlet at 5.54 ppm for the CH group, a singlet at 6.36 ppm for the CH group of the 4H-pyrone moiety, a broad signal at 6.60 ppm for two NH protons of urea group, a multiplets at 7.16–7.27 ppm and a doublet of doublet signals at 7.36 ppm for the aromatic protons clearly confirmed the structure. The number of peaks in 13C-NMR spectrum and the total numbers of carbon atoms for UPC structure confirmed these 24 signals as described in Table 1.
3.2 Modification of SiO2 Nanoparticles
FT-IR spectrum of the SiO2 nanoparticles showed two characteristic peaks at about 3700–3200 and 1590 cm−1, which could be attributed to the stretching and bending vibrations of hydroxyl groups on the surface of SiO2 nanoparticles. Also, an absorption band below 700 cm−1 was appropriate to Si–O band. The sharp bands around 780, 554 and 450 cm−1 were caused by various lattice vibrations associated to the connected silica oxide structures. In the FT-IR spectrum of the surface modified SiO2 nanoparticles (OMSN) the unique absorption band of Si–O–Si appeared at 1086 cm−1 and absorption peaks related to the modifier were obviously observed in the formation of the Si–O–Si linkage after hydrolysis and condensation. Furthermore, treated SiO2 by UPC illustrated the peak at 3424 cm−1 related to the OH stretching, and peak at 2960 cm−1 related to the stretching vibration of CH units. Also a few low intensity peaks from 2000 to 1500 cm−1 were observed. The entire signals of the synthetic coupling agent were detected by slight displacements in wavenumber from the parent structure.
The weight of the organic structure grafted onto 100 g of silica nanoparticles was defined as the grafting percentage and was calculated from the thermogravimetry analysis (TGA). For OMSN, the grafting percentage were obtained by using thermogram by inspection of the residual weight of the prepared OMSN at 200 and 650 °C. TGA curve of the pure nanopartilces do not show any significant weight loss with temperature before 150 °C. But in the thermogram of silica nanoparticles weight loss below 130 °C was observed. It can be attributed to evaporation of the residual moisture in the specimen [29]. For OMSN, weight loss at about 400–470 °C and a maximum degradation rate at 430 °C was attributed to the degradation of UPC linked onto the surface of the pure silica nanoparticles. Grafting amount of the UPC in the OMSN surface was calculated to be 6.5%. TG analysis of OMSN also showed the weight decreases gradually over 350 °C and the loss is about 5% at about 500 °C, which can be attributed to the thermal decomposition of UPC at higher temperatures due to the presence of chlorine atom and aromaric ring in the UPC structure.
PVA/SiO2 hybrid films were synthesized according to the synthesis pathway reported in our previous article [22]. The hydrogen bonding and van der Waals forces between polymer segments and OMSN was occurred and its chemical illustration was shown in Scheme 2. OMSN was introduced in PVA matrix with different nano contents containing 2, 5, 10, and 15 wt%.

Interactions of PVA and modified SiO2 nanoparticles in nanocomposites
The structural compositions of the prepared nanocomposites were examined by FT-IR and UV–Vis spectroscopy. Figure 1b–e shows the FT-IR spectra of prepared samples. In the spectrum of neat PVA (Fig. 1a), the distinctive absorption peak at around 3500–3400 cm−1 is attributed to the O–H stretching vibration of hydroxyl groups. The absorption peak in the region of 2941 cm−1 is related to the stretching vibrations of CH and CH2 groups and also the peak at 1428 cm−1 may be corresponding to the C–H bending vibration in the PVA chains. Furthermore, a strong absorption peak at 1741 cm−1 is associated to the carbonyl functional units of the residual unreacted acetate groups presented after production process in PVA. The absorption peak at 1094 cm−1 is associated to the C–O stretching vibrations related to the PVA matrix. In the FT-IR spectra of SiO2/PVA nanocomposites, the characteristic absorbing peaks related to the vibration of Si–O–Si groups of nanoparticles are seemed to be observed at around 800–450 cm−1. Conversely, after the addition of nanostructure particles, C=O vibrational stretching peaks shifted to the higher wavenumbers that can be due to the enrichment in the number of the physical interaction between nanocomposite components. This suggests that the existence of hydrogen bonding interactions between the PVA matrix and silica nanoparticles has been improved. The prepared nanocomposites exhibited the absorption peaks at about 1780–1720 cm−1 related to the different C=O stretching of ester, carbonyl and urea linkages. The C–N stretching vibration of urea units appeared at 1382 cm−1. The existence of the strong absorption peaks at 1776 and 1724 cm−1 related to the UPC functional units on the OMSN and the characteristic vibrations peaks related to the self-silica nanoparticles were not clearly detectable.

FT-IR spectra of a PVA, b PVA/SiO2 (2 wt%), c PVA/SiO2 (5 wt%), d PVA/SiO2 (10 wt%), e PVA/SiO2 (15 wt%)
UV–Vis spectra of PVA/SiO2 nanocomposite films with different amounts of OMSN dispersed in the polymeric matrices are shown in Fig. 2. The UPC solution has an absorption wavelength around 225 and 275 nm. The PVA has no obvious absorption at 200–600 nm. The PVA/silica nanocomposites displayed an absorption peak at the wavelength around 250–300 nm. The absorption intensities for UPC linked SiO2 nanoparticles clearly displayed the chemical attachment of UPC units on to the silica spheres and the intensities of peaks increased gradually as a function of nanoparticles contents. Increasing of the OMSN contents in the nanocomposites caused to strength in peak intensities. The prepared PVA nanocomposites absorb UV light at around 200–300 nm. The nanocomposites maximum absorptions are found to be at 256 nm. According to the spectra, PVA/SiO2 nanocomposites with higher loading level of nano-materials showed the high absorption peaks between 200 and 350 nm. The results confirm that the contents of OMSNs which contain UV active chromophores have noticeable effect on the absorption of PVA nanocomposites.

UV absorption spectra of the PVA/SiO2 nanocomposites
3.3 Morphology of Nanocomposite Film
Figure 3 shows the FE-SEM images of the prepared films by using the OMSN. It is apparent that in the FE-SEM images, narrow size distribution, homogeneous shapes and separated particles for OMSNs were clearly observed. By investigation of the prepared nanocomposites, it was revealed that the OMSN was finely dispersed in the matrix, whereas the dispersions of the silica nanoparticles in matrix were relatively constant. The organic structure linked to the SiO2 nanoparticles showed better compatibility with the PVAmatrix. However, the unique structure of PVA was not damaged by modified SiO2. The analysis of the FE-SEM images showed that the OMSNs with dimension of 23–86 nm were embedded in PVA matrix. In Fig. 3a, the diameter of silica nanoparticles was alternating from 60 to 90 nm. These nanoparticles have uniform distribution into the PVA matrix according to the FE-SEM images of PVA/silica nanocomposites. From 2 to 15 wt% samples, no obvious aggregations of nanoparticles were observed in the FE-SEM images due to the better performance of coupling agent characteristics. The comparison between parts b and d of Fig. 3 showed that by enrichment in SiO2 contents in nanocomposites, the sizes of inorganic domains were increased. The presence of different functional groups such as hydroxyl, urea, and carbonyl units on the surface of modified SiO2 nanoparticles provided the hydrogen bonding or physical interactions to form building blocks to increase the sizes of the nanoparticles. This caused to create the agglomerated nanoparticles. Therefore the sizes of nanoparticles in nanocomposite containing 15 wt% modified nanosilica have been enlarged.

FE-SEM micrographs of nanocomposites (2–15 wt%)
The AFM image provides information about surface morphologies by using two- and three-dimensions. AFM images were provided in outlay with around 3 × 3 μm2 scan area from the original images. Figure 4 shows two, and three-dimension AFM images of surface topography of PVA matrix. The images b, c and d showed the two dimensional topographic images of Fig. 4. The Fig. 4c, d illustrated the canny edge detection presentation and the step edge detection presentation of the Fig. 4a.

Two and three dimensional AFM topography images of PVA matrix
Also Fig. 5a, b displayed pictures related to the three dimensional topographic images for nanocomposite 10 wt%. The Fig. 5d, f demonstrated the canny edge detection and the step edge detection presentations of the Fig. 5b. It comes out from three-dimensional 3 × 3 μm2 outline for nanocomposite 10 wt%, an unsymetrical structure was observed and also the maximum particles projected from the surface was around 105 nm. It appeared that by insertion of the surface treated silica nanoparticles in PVA matrix the projected particles due to the residual of silica nanoparticles on the surface were increased. The good dispersion of silica nanopartiles in polymer matrix could be confirmed by the observation of the micrographs from FE-SEM analysis results.

Two and three dimensional AFM topography images of NC 10 wt%
In the length of 3 μm, five different lines with sizes smaller than 2 μm were drawn in distinct measurement length. By inspection of the AFM images, it was seen that the irregular dispersal of peaks and valleys related to the residual of nanoparticles on the surfaces of polymer matrix for nanocomposite 10 wt% were observed. According to the observation of AFM data the maximum profile for the evaluation length was detected to be smaller than 60 nm (Fig. 6).

Two dimensional images with five different gates
Also through the measuring line of selected AFM image of nanocomposite 10 wt%, the graphs related to the mean values (b), minimum values (c), maximum values (d), RMS value (e), and height distribution profile (f) were obtained and shown in collected Fig. 7. Figure 7a showed that the mean values of protruded particles from the surface of nanocomposite 10 wt% were below than 30 nm. The minimum profile graph showed that in many positions the films were very smooth. Section (d) of Fig. 7 illustrates the height of projected particles profiles on the surface of nanocomposite. Also Fig. 7e, f clearly showed the characteristics of protruded particles on the surfaces of nanocomposite 10 wt%.

Two dimensional AFM topography image of NC 10 wt% (a), mean values (b), minimum values (c), maximum values (d), RMS value (e), and height distribution profile (f)
Geometric analysis can provide quantitative values for investigation of the surface morphology and alteration of the structures presented in nanocomposites. Fractal geometry can be used to quantitatively describe irregular shapes and surfaces in nanocomposite materials. The basic quantitative component of fractal geometry, the fractal dimension D, is a non-integer number with a Euclidean and non-Euclidean component. The Euclidean component describes the topological dimension of the object, i.e., 1 for a line, 2 for an area or surface, and 3 for a volume. The non-Euclidean component, called the fractal dimensional increment, D*, describes the level of irregularity of the object from its Euclidean geometry. The fractal dimensional increment ranges from 0 to 1, with the level of irregularity increasing as the increment increases. Several algorithms have been used to regulate the value of fractal dimension D constructed on the AFM data. In order to study the fractal features of silica nanocomposites, to calculate D from the AFM data, a Gwyddion software package was used. Gwyddion is a free and open source software program for SPM data visualization and analysis [30]. Gwyddion uses four methods to calculate D from AFM data: (1) cube counting, (2) triangulation, (3) partitioning, and (4) power spectrum distribution (Fig. 8). The average D* values for the nanocomposite 10 wt% based on cube counting, triangulation, variance, and power spectrum methods were 0.17, 0.22, 0.26, and 0.13 respectively. Fractal analysis has shown that all samples exhibit a self-similar character, which is concern to the fabrication route and is temperately sensitive to the nanoparticles content in nanocomposites. Figure 8 illustrated the (a) power spectrum, (b) triangulation, (c) cube counting and (d) partitioning, and linear fit graphs for nanocomposite 10 wt%. Based on AFM and SEM observations, the surface roughness of prepared nanocomposites was related to the surface morphology, to the nanofiller content and to the average filler size. Nanocomposite comprised 2 wt% nanoparticles exhibited the smoothest surface, followed by 5, 10, and 15 wt% samples. Positive correlation between the average nanoparticle size, the nanoparticle content and the surface roughness suggest that using more nanoparticles in the preparation of nanocomposite deteriorate the surface texture. Based on the illustrated mechanism in Scheme 2, by increase in the interaction between functional units on the surface of modified nanoparticles the surface roughness of nanocomposite was increased.

Fractal analysis based on the a power spectrum, b triangulation, c cube counting and d variance methods, and linear fit graphs for nanocomposite 8 wt%
We have inspected the thermal stability of PVA/SiO2 nanocomposites by the use of thermogravimetric analyses(TGA), in which the weight loss of these nanocomposites was measured by raising the temperature around 700 °C at heating rate 10 °C/min under nitrogen atmospheric conditions. Figure 9 explained the TG/DTG curves of the nanocomposite 10 wt% silica from room temperature to 700 °C. Thermal stability evaluation of the nanocomposite 10 wt% silica was measured based on the degradation temperature of 5% weight loss (T5) and the degradation temperature of 10% weight loss (T10) of the nanocomposite. The value of T5 for prepared sample 10 wt% nanocomposite was measured around 260 °C and T10 value was estimated above 289 °C, and the residual weight for this nanocomposite at 700 °C ranged in 6% in nitrogen atmosphere. According to TGA results, the thermal degradation behavior of nanocomposites with different percentages of modified silica showed one minor and three major weight loss steps when heated from room temperature to 700 °C. The minor decomposition step starts at around 75–100 °C and the reason for the weight loss may be caused by the vaporization of the adsorbed water and trapped water owing to the interactions between PVA and water. The first major decrease in weight in the second step was related to the decomposition of alkyl units present in the structure of the coupling agent and main chain decomposition (at about 317 °C). The second major weight loss step for the nanocomposite 10 wt% occurs at about 360 °C. Decomposition of the aromatic component of coupling agent is expected to take place between 450 and 530 °C (third major). This improvement in thermal stability of the PVA network was related to the presence of organic and inorganic moieties in the structure of nanocomposites. TGA analysis result of pure PVA showed three steps weight loss patterns. The first step was perceived at 76 °C with 3% weight loss, related to the evaporation of adsorbed water in PVA chains. The value of T5 for PVA sample was measured around 220 °C. The second step at 220–280 °C with 80% weight loss could be ascribed to the elimination of water or the polymer dehydration and formation of a polyacetylene-like structure. The third step at 400–550 °C with 92% weight loss was described to the decomposition of main chain of PVA. This enhancement in the thermal stability is caused by the presence of organic silicone nanoparticles, which acts as a barrier to maximize the heat insulation and minimize the permeability of volatile degradation products in the nanocomposites. Based on these results, the degradation process will be slowed and decomposition will happen at higher temperatures. Char yield (CR) can be used as criteria for evaluating limiting oxygen index (LOI) of the polymers in accordance with Van Krevelen and Hoftyzer equation [31], LOI = 17.5 + 0.4CR. In general, when the LOI of a material is higher than 26%, it is considered to be flammable. For these nanocomposites, LOI values were calculated based on their char yields at 700 °C. According to Table 2, thermal stability of the nanocomposites in N2 appeared in the range 472–511 °C in terms of T10 values.

TGA/DTG thermograph of PVA/SiO2 NC (10 wt%) under the nitrogen atmosphere at heating rate of 10 °C/min
4 Conclusions
In this study the surface of silica nanoparticles were modified with 4-H pyran derived structure as organo silicone modifier. FT-IR and TGA analyses confirmed the grafting of 4-H pyrane derived structure onto the surface of silica nanoparticles. FE-SEM approved the well dispersion of nanoparticles after the surface modification process. Then, modified SiO2 was introduced into the PVA matrix and PVA/SiO2 nanocomposites were obtained. These nanocomposites showed good thermal stability. Results based on the FE-SEM images showed that the nanostructured materials with uniform shape and size were dispersed in polymeric matrix. The hydrogen bonding interactions can be formed between the functional units of PVA and the hydroxyl and other functional units of 4-H pyrane derived structure presented on the surface of modified silica. The thermal, morphological and optical properties of the PVA/silica nanocomposites were investigated. The obtained results showed enhancement of PVA properties in comparison to the pristine PVA. The morphologies of nanocomposites structures are varied from original PVA and uniform dispersion of nanofillers in the PVA matrix were observed. The thermal analysis results indicated that the incorporation of the nanofillers was enhanced the thermal stability of the PVA nanocomposites films after using modified nano silica. AFM analysis outcomes revealed particular places containing silica nanoparticles from the surface of nanocomposites. The UV–Vis spectra showed the existence of 4-H Pyrane derivative as chromosphere in the structure of nanocomposites.
References
J. Zhu, Q. Wang, M.-X. Wang, Multicomponent reactions in organic synthesis. (Wiley-VCH, Weinheim, 2015)
G. Brahmachari, Green synthetic approaches for biologically relevant heterocycles. (Elsevier, Amsterdam, 2015)
Y. Li, B. Zhao, B. Du, Q. Jiang, X. Wang, C. Cao, Tetrahedron Lett. 54, 227 (2013)
R. Pagadala, S. Maddila, S.B. Jonnalagadda, Org. Prep. Proced. Int. 46, 261 (2014)
M. Obst, T. Heinze, Macromol. Mater. Eng. 301, 65 (2016)
M.D. Aytemir, U. Calis, M. Ozalp, Arch. Pharm. Pharm. Med. Chem. 337, 281 (2004)
A.R. Saundane, K. Vijaykumar, A.V. Vaijinath, Bioorg. Med. Chem. Lett. 23, 1978 (2013)
H.-I. Hsiang, Y.-L. Chang, C.-Y. Chen, F.-S. Yen, Mat. Chem. Phys. 120, 476 (2010)
Y. Oikawa, T. Saito, S. Idomukai, T. Tanaka, M. Nishida, H. Sawada, J. Fluorine Chem. 177, 70 (2015)
M. Hatami, B.F. Azarkar, M. Qandalee, H. Hasanabadi, Polym. Eng. Sci. 55, 2339 (2015)
M. Taghavi, M. Ghaemy, S.M.A. Nasab, M. Hassanzadeh, J. Polym. Res. 22, 1 (2015)
X. Wang, P. Wang, Y. Jiang, Q. Su, J. Zheng, Compos. Sci. Technol. 104, 1 (2014)
L.-Y. Meng, S. Han, N. Jiang, W. Meng, Mat. Chem. Phys. 148, 798 (2014)
L.H. Manchini, C.L. Espozito, Nanocomposites: Preparation, properties, and Performance. (Nova Science Publisher, New York, 2008)
A.T. Mane, S.T. Navale, R.S. Mane, M. Naushad, V.B. Patil, Prog. Org. Coat. 87, 88 (2015)
S. Mallakpour, M. Hatami, A.A. Ensafi, H. Karimi Maleh, J. Solid State Electrochem. 115, 2053 (2011)
S. Yazdani, M. Hatami, S.M. Vahdat, Turk. J. Chem. 38, 388 (2014)
R. Marandi, M.E. Olya, B. Vahid, M. Khosravi, M. Hatami, Environ. Eng. Sci. 29, 957 (2012)
S. Azizi, M.B. Ahmad, M.Z. Hussein, N.A. Ibrahim, F. Namvar, Int. J. Nanomedicine 9, 1909 (2014)
S. Asghari, S. Ramezani, M. Ahmadipour, M. Hatami, Des. Monomers Polym. 13, 73 (2011)
R. Shahabadi, M. Abdollahi, A. Sharif, Int. J. Hydrogen Energy 40, 3749 (2015)
S. Asghari, M. Hatami, M. Ahmadipour, Polym. Plast. Technol. Eng. 54, 192 (2015)
S. Mallakpour, A. Abdolmaleki, Z. khalesi, S. Borandeh, Polymer 81, 140 (2015)
V. Mittal, Advanced polymer nanoparticles: synthesis and surface modification. (Taylor and Francis, London, 2011)
S. Mallakpour, L. Mani, Prog. Org. Coat. 78, 35 (2015)
M. Abdollahi, M. Rouhani, M. Hemmati, P. Salarizadeh, Polym. Int. 62, 713 (2013)
S. Asghari, R. Baharfar, M. Alimi, M. Ahmadipour, M. Mohseni, Monatsh Chem. 145, 1337 (2014)
A.M. Buckley, M. Greenblatt, J. Chem. Ed. 71, 599 (1994)
R. Goyal, S.K. Tripathi, E. Vazquez, P. Kumar, K.C. Gupta, Biomacromolecules 13, 73 (2011)
D. Nečas, P. Klapetek, Gwyddion - Free SPM (AFM, SNOM/NSOM, STM, MFM, …) data analysis software. Gwyddion - Free SPM (AFM, SNOM/NSOM, STM, MFM, …) data analysis software (2009), http://gwyddion.net/. Accessed 01 Feb 2016
D.W. Van Krevelen, P.J. Hoftyzer, Properties of Polymers, 3rd edn. (Elsevier, Amsterdam, 1976)
Acknowledgements
We wish to express our thankfulness to the Research Affairs Division University of Mazandaran, Babolsar, and also University of Bonab, Bonab for partial financial support. Further financial support from Iran Nanotechnology Initiative Council (INIC) is appreciatively acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hatami, M., Ahmadipour, M. & Asghari, S. Heterocyclic Grafting Functionalization of Silica Nanoparticles: Fabrication, Morphological Investigation and Application for PVA Nanocomposites. J Inorg Organomet Polym 27, 1072–1083 (2017). https://doi.org/10.1007/s10904-017-0557-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-017-0557-1