
Abstract
Two palladium(II) nitroaryl complexes trans-[bromo(p-nitrophenyl)bis(triphenylphosphine)palladium(II)] 1 and trans-[bromo(2,4-dinitrophenyl)bis(triphenylphosphine)palladium(II)] 2 have been synthesized. The complexes were characterized by FTIR and NMR (1H, 13C and 31P) spectroscopy and elemental analysis. The molecular structure of complex 2, as confirmed by X-ray crystallography, reveals that the Pd atom and its neighboring groups (two PPh3, Br and phenylene group) lie in a slightly distorted square plane. In the UV–Vis spectra of the complexes 1 and 2, the palladium to aryl charge transfer bands were observed. The emission peaks from the singlet excited states (S1 → S0) were observed in the photoluminescence spectra of the complexes. The thermal stability of the complexes has been studied by thermal gravimetric analysis (TGA). TGA data showed that both complexes are thermally stable up to 200 °C, and complex 1 is more stable than 2. The catalytic efficiency of the new palladium(II) complexes was studied as demonstrated using the Sonogashira coupling reactions with good yields. The experimental results suggest that the Sonogashira coupling reactions can be performed at moderate temperature (50 °C) using these new palladium(II) complexes as catalysts.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Remarkable progress has been made in the area of palladium(II) complexes due to their promising properties in applied sciences, mostly owing to the high efficiency as homogeneous catalysts in a variety of coupling reactions in organic synthesis [1]. Such palladium(II) catalysts have been widely applied to prepare biologically-active molecules, natural products, conducting polymers and macrocycles with acetylene links [2, 3]. Palladium(II) catalysis has also gained widespread use in industrial and academic synthetic chemistry laboratories as a powerful methodology for the formation of C–C and C-heteroatom bonds [4]. Besides, the palladium-catalyzed cross-coupling between sp2-hybridized carbon atoms (C(sp2)) of aryl, heteroaryl, and vinyl halides and sp-hybridized carbon atoms of terminal acetylenes (C(sp)) pertains to the family of modern and extremely powerful synthetic methods for the synthesis of important organic intermediates and products. Collectively, the seminal works by Stephens and Castro [5], Dieck and Heck [6], Cassar [7] and Sonogashira, Tohda, and Hagihara [8] have initiated an outstanding number of studies in the fields of organic chemistry, organometallic chemistry and materials science [9].
Many Pd-catalyzed reactions were developed over the past three decades, and the growing number of scientific publications in this area shows its high demand. There are two main reasons for the growing interest in this field. The first is that this catalyst system provides a simple and feasible method for creating carbon–carbon and carbon–nitrogen bonds under sustainable conditions with excellent yield, and at the same time, due to mild reaction conditions, it is highly tolerable by many functional groups [10]. The most commonly used catalytic systems for this transformation include PdCl2(PPh3)2, PdCl2/PPh3, and Pd(PPh3)4 together with CuI as the co-catalyst and a large amount of amines as the solvents or co-solvents [11–13]. In this connection, the palladium-catalyzed coupling reactions reported by Sonogashira-(Hagihara) [8, 14] are recognized as a powerful and reliable synthetic method for the formation sp2C–spC bonds in substituted or non-substituted acetylenes. The unique properties of acetylenic arrays continue to attract considerable interest with research in nonlinear optical electronic devices and in natural product synthesis [15, 16].
In the present work, we report the synthesis of two new palladium(II) complexes [bromo(p-nitrophenyl)bis(triphenylphosphine)palladium(II)] 1 and trans[bromo(2,4-dinitrophenyl)bis(triphenylphosphine)palladium(II)] 2 which furnish a highly active catalyst for Sonogashira coupling, providing a mild, efficient and general method for the reactions of nitroaryl bromides with different terminal acetylenes.
2 Experimental
2.1 Materials and Methods
All the reagents used in this research work were analytical grade and chemically pure. The solvents were dried according to the standard procedures [17]. Palladium(II) chloride (palladium 59.9 %), triphenylphosphine (99 %) and hydrazine hydrate (99 %) were obtained from Johnson Metthey, Acros and BDH, respectively. p-Nitrobromobenzene (99 %), 2,4-nitrobromobenzene (99 %), 2-methyl-3-butyn-2-ol (98 %), phenylacetylene (98 %), tolylacetylene (98 %) and copper(I) iodide (99.999 %) were purchased from Sigma Aldrich. Toluene (≥99 %), DMSO (≥99 %), n-hexane (≥95 %), diisopropylamine (99 %), piperidine (99 %) and trimethylsilylacetylene (98 %) were obtained from MERCK. Dichloromethane (99.8 %) was obtained from RCI labscan, Thailand. The FT-IR spectral measurements were done using KBr as pellets on Shimadzu IR spectrophotometer (Prestige-21) in the range 4000–400 cm−1. Elemental analysis was performed using Vario Micro Cube elemental analyzer. All the NMR samples were prepared as CDCl3 solutions, and standard 1H- and 13C NMR spectra were recorded on Bruker NMR spectrometer at 400 and 101 MHz, respectively. 1H- and 13C NMR spectra were referenced to residual CHCl3 of the solvent. The 31P NMR spectra of the complexes were obtained at room temperature using o-phosphoric acid as a reference. Emission spectra were recorded on Shimadzu RF-5301PC spectrofluorophotometer, and TGA data were recorded on Shimadzu TGA-50 thermogravimetric analyzer. Single crystal of complex 2, suitable for X-ray crystallographic analyses, was chosen and mounted on a glass fiber using epoxy resin. Crystal data and other experimental details are summarized in Table 2. The diffraction experiments were carried out at 173 K on a Bruker APEX-II CCD area-detector diffractometer using graphite-monochromated Mo-K α radiation (λ = 0.71073 Å). The collected frames were processed with the software SAINT [18], and an absorption correction (SADABS) was applied to the collected reflections [19]. The structures were solved by direct methods, and expanded by difference Fourier syntheses using the software SHELTXL [20]. Structure refinements were made on F2 by the full-matrix least-squares technique. In each case, all the non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were placed in their ideal positions but not refined (CCDC 1478703).
2.2 Syntheses
2.2.1 Tetrakis(triphenylphosphine)palladium(0)
The tetrakis(triphenylphosphine)palladium(0) was synthesized according to the previously reported procedure [21]. PdCl2 (0.100 g, 0.565 mmol) and triphenylphosphine (0.745 g, 2.82 mmol) were added to 15 mL of dry and degassed DMSO. Then the reaction mixture was heated at 140 °C for 0.5 h. The resulting solution was cooled down to room temperature, and hydrazine hydrate (0.113 mL, 3.62 mmol) was added to the solution with vigorous starring. During stirring a yellow precipitate was formed which was filtered and washed with dry ethanol and ether, and dried under vacuum. The pure product was isolated as a yellow solid in 93 % yield.
2.2.2 Trans-[bromo(p-nitrophenyl)bis(triphenylphosphine)palladium(II) Complex 1
Tetrakis(triphenylphosphine)palladium(0) (0.240 g, 0.207 mmol) and 4-nitrobromobenzene (0.053 g, 0.249 mmol) were added to the freshly dried and degassed toluene (15 mL). The reaction was subsequently refluxed for 5.5 h [22]. The solvent was removed under reduced pressure, and the residue was subjected to silica column chromatography (3/1: dichloromethane/n-hexane, v/v). The pale yellow product was dried under vacuum and isolated in 93 % yield.
FTIR: 3054, 2923, 1588, 1557, 1503, 1480, 1464, 1433, 1348, 1310, 1178, 1094, 1070, 1047, 847, 823, 738, 692, 507, 453, 422 cm−1; 1H NMR (δ ppm): 6.86 (d, 2H, 3 J = 8.4 Hz), 7.01 (d, 2H, 3 J = 8.4 Hz),7.54 (dd, 3 J = 6 Hz, 12H), 7.25 (t, 3 J = 7.4 Hz, 12H), 7.34 (t, 3 J = 7.2 Hz, 6H); 13C (δ ppm), 174.16, 136.10, 120.80, 143.74, 130.75 (t, 1 J C−P = 23.55 Hz), 134.67 (t, 2 J C−P = 6.1 Hz), 128.07 (t, 3 J C−P = 4.75 Hz), 130.25 (s); 13C NMR (135 DEPT) (δ ppm): 136.10, 120.80, 143.74, 134.67, 128.07, 130.25; 31P NMR (δ ppm): 24.98 (s) Anal. Calc. for C42H34NO2BrPd: C 60.56; H 4.11; N 1.68. Found: C 61.14; H 3.67; N 1.67 %.
2.2.3 Trans-[bromo(2,4-dinitrophenyl)bis(triphenylphosphine)palladium(II) Complex 2
As for the formation of 1, but using 2,4-dinitrobromobenzene (0.200 g 0.173 mmol) the reaction gave trans-[bromo(2,4-dinitrophenyl)bis (triphenylphosphine)palladium(II) 2 as a pale yellow solid in 40 % yield.
FTIR (KBr): 3057, 2924, 1577, 1508, 1481, 1435, 1340, 1330, 1232, 1188, 1159, 1134, 1097, 1072, 1031, 999, 908, 833, 734, 692, 522, 511, 493, 426 cm−1; 1H NMR (δ ppm): 7.44 (dd, 1H, 3 J = 3 Hz), 7.65 (d, 1H, 3 J = 8.4 Hz), 7.90 (s), 7.56 (dd, 13H, 3 J = 3 Hz), 7.26 (t, 13H, 3 J = 7.4 Hz), 7.35 (t, 7H, 3 J = 7.4 Hz); 13C NMR (δ ppm): 175.93, 152.24, 120.11, 144.45, 123.35, 137.77, 130.15 (t, 1 J C−P = 23.8 Hz), 134.42 (t, 2 J C−-P = 6.2 Hz), 128.22 (t, 3 J C−P = 5 Hz), 130.47 (s); 13C NMR (135 DEPT) (δ ppm): 120.11, 123.35, 137.77, 134.42, 128.22, 130.47; 31P NMR (δ ppm), 22.65 (s), Anal: Calc. for C42H33N2O4BrPd.0.5 n-hexane: C 58.68; H, 4.38; N, 3.04. Found: C 58.49; H 3.92; N 3.09 %.
2.3 General Procedure for Sonogashira Coupling Reactions
Sonogashira coupling reactions of 4-nitrobromobenzene with different terminal acetylenes, using palladium(II) complex 1 or 2 as the catalyst, was carried out following the general procedure given below [8, 14]. 4-Nitrobromobenzene and acetylene were added to a freshly dried and degassed diisopropylammine (25 mL) or piperidine (10 mL) in the presence of complex 1 (2 mol %), PPh3 (4 mol %) and CuI (2 mol %) in an ice bath with a positive flow of nitrogen. After 30 min, the reaction temperature was raised to 50 °C, and the reaction was stirred for 20 h. Once the reaction was complete, the mixture was filtered, and the solvent was removed by a rotary evaporator. Then the residue was extracted by diethyl ether, and the ether solution was washed with 10 % aqueous HCl, saturated aqueous NaHCO3 and brine solution, and dried over anhydrous MgSO4. Then the solvent was removed under reduced pressure and the residue was subjected to silica column chromatography using dichloromethane/n-hexane as eluent. The products were characterized by comparing spectroscopic data of the authentic samples reported in the literature [23–26].
3 Results and Discussion
3.1 Syntheses
Palladium(II) complexes of trans-[bromo(p-nitrophenyl)bis(triphenylphosphine)palladium(II) 1 was formed in 93 % yield by the oxidative addition of 2-nitrobromobenzene to tetrakis(triphenylphosphine)palladium(0) in toluene (Scheme 1). Trans-[bromo(2,4-dinitrophenyl)bis(triphenylphosphine)palladium(II) 2 was formed similarly.

Synthesis of trans-[bromo(p-nitrophenyl)bis(triphenylphosphine)palladium(II)
The analytical data of the palladium(II) complexes 1 and 2 are in good agreement with the calculated values, thus confirming the proposed mononuclear composition for both complexes. The new complexes are stable at room temperature, and soluble in common organic solvents such as dichloromethane, ethanol, methanol and DMSO.
3.2 Characterization
The structures of complexes 1 and 2 were confirmed by FT-IR and multinuclear NMR (1H, 13C and 31P) spectroscopy.
3.2.1 FT-IR Spectroscopy
The FT-IR spectra of both reactants were compared with the spectra of the synthesized products in order to confirm the formation of new complexes 1 and 2. A band was observed at 425 cm−1 for both complexes, which is the characteristic peak of Pd-Br bond [27]. The peaks displayed at 1514 and 1346 cm−1 in the IR spectrum of p-nitrobromobenzene were assigned to the presence of aromatic nitro group which had been shifted to the lower frequencies at 1508 and 1340 cm−1 in complex 1. The peaks observed at 1579 and 1471 cm−1 due to the presence of aromatic C=C bond also shifted to 1586 and 1479 cm−1, indicating the formation of complex 1. Similar IR spectral features were also observed for complex 2, which confirmed the formation of such complex.
3.2.2 NMR Spectroscopy
Figure 1 shows the 1H NMR spectrum of trans-[bromo(p-nitrophenyl)bis (triphenylphosphine)palladium(II)] (complex 1), where the proton signals arise from the phenyl ring of the triphenylphosphine and nitrobenzene. Complex 1 displayed two doublets at 6.86 and 7.01 ppm due to the four Ha and Hb protons of nitrobenzene. Two doublets of doublet at 7.54 ppm, one triplet at 7.25 ppm and one triplet at 7.34 ppm are attributed to the protons attached to the ortho (12 protons), para (12 protons) and meta (6 protons) positions of the phenyl ring attached to the phosphorus atom (Fig. 1) [28].

1H NMR spectra of trans-[bromo(p-nitrophenyl)bis(triphenylphosphine)palladium(II)]
The 1H NMR spectrum of trans-[bromo(2,4-dinitrophenyl)bis (triphenylphosphine)palladium(II)] (complex 2) also displayed similar signals in the expected region (Table 1). The 13C NMR spectra of the palladium(II) complexes showed peaks at 120-176 ppm region due to the presence of aromatic carbons. Complex 1 displayed five signals at 174.16, 136.10, 120.80, 143.74, and 130.25 for C-1, C-2, C-3, C-4 and C-8 carbon. Here, C-5, C-6 and C-7 carbon appear as a triplet at 130.75, 134.67 and 128.07 ppm (3 J pc = 23.55, 6.1 and 4.75 Hz) due to the coupling of the carbon with the phosphorus atom. The 13C NMR (135 DEPT) spectra (Fig. 2) did not show any signal for ipso carbon (C-1, C-4 and C-5) which confirmed the presence of nonhydrogenated carbon. Trans-[bromo(2,4-dinitrophenyl)bis (triphenylphosphine)palladium(II)] (complex 2) also displayed similar signals in the 13C NMR spectra (Table 1).

13C NMR 135 DEPT spectra of trans-[bromo(p-nitrophenyl)bis triphenylphosphine)palladium(II)]
The 31P NMR spectra were recorded for complexes 1 and 2 in order to confirm the presence of triphenylphosphine group and its geometry in the new palladium(II) complexes. The 31P NMR spectrum clearly showed a signal at 24.98 ppm for complex 1 (Fig. 3a), which is attributed to the bonded triphenylphosphine ligand on palladium. The chemical shift in the 31P NMR spectrum of the starting complex tetrakis(triphenylphosphine)palladium(0) at ambient temperature in DMF-d 7 was δ 17.1 ppm which is in the downfield side of the spectrum compared to the δ value of complex 1. Complex 2 also displayed similar signals in the 31P NMR spectrum.

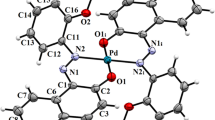
A perspective view of complex 2, with the thermal ellipsoids shown at the 50 % probability level
3.3 Single-Crystal X-ray Crystallography
The molecular structure of complex 2, as confirmed by X-ray crystallography, is shown in Fig. 3, and Table 3 lists the bond lengths (Table 2) and bond angles. In complex 2, the Pd atom and its neighboring atoms, P(1), P(2), Br and C(1) lie in a slightly distorted square plane. A least-squares plane calculation reveals the planarity of the P(2)-P(1)-C(1)-Br core (largest deviation 0.08 Å). The Pd–O bond distance of 2.829 Å indicates no bonding interaction between the nitrogen atom and the palladium center. Within the dinitrobenzene and triphenylphosphine ligands, the geometries are consistent with a significant partial double bond character in the C–C bonds. The Pd–C [1.992(2) Å] and Pd–Br [2.4913(3) Å] lengths for 2 are in good agreement with the reported values [29, 30].
In the complex, the Pd(1)–P(1) bond length [2.3259(7) Å] is slightly shorter than that of Pd(1)–P(2) [2.3315(7) Å]. Both P atoms of the triphenylphosphine ligands have similar distorted tetrahedral geometry. The P(2)-Pd(1)-Br(1) angle is less distorted from 90o than the P(1)-Pd(1)-C(1) and P(2)-Pd(1)-C(1) angles. The P(1)-Pd(1)-Br(1) angle is greater than 90o in the complex due to the steric effect of the dinitrophenyl ligand. The N–O bond distances (average = 1.222 Å) are almost similar in all the nitro groups of the dinitrophenyl rings indicating the partial double bond character.
3.4 Electronic Spectroscopy
The UV–Vis spectra of the new complexes 1 and 2 have been recorded in CH2Cl2 solution in the region 800–200 nm (Fig. 4). The absorption band for complex 1 at λmax 330 nm is assigned to a spin-allowed 4d (Pd) → (L) (MLCT) transition [30]. Complex 2 also displayed similar absorption band. The emission spectra of 1 and 2, at room temperature, in CH2Cl2, upon excitation at 330 nm are shown in Fig. 4. For complex 1, the luminescence peaking at 404 nm was assigned to the emission from singlet excited state (S1 → S0) with small Stokes shift (74 nm) between the absorption and emission bands. It is ascribed to the metal-perturbed intraligand excited states [31]. Complex 2 also displayed similar spectral features.

Normalized absorption and emission spectra of complexes 1 and 2 in CH2Cl2
3.5 Thermal Studies
The thermal stability of complexes 1 and 2 was studied by thermogravimetric analysis (TGA) under a nitrogen atmosphere. The TGA curves of complexes 1 and 2 are presented in Fig. 5. A single step decomposition process for 1 and 2 is observed for both complexes in the range of temperature 288–321 °C and 244–339 °C, respectively. Thus, these two complexes show different thermal stability behaviors, and the complex 1 is more stable than complex 2. For the complex 1, the percentage of weight loss was 61.5 %, while for complex 2 the weight loss was 75.0 %. The total % weight loss of the complexes may correspond to the loss of the respective ligands [32].

TGA curves of complexes 1 and 2
3.6 Catalytic Activity
3.6.1 Sonogashira Coupling Reactions of 4-nitrobromobenzene with Different Terminal Acetylenes
The newly formed palladium(II) complexes were employed as the catalyst (complexes 1 and 2 are termed as catalysts 1 and 2, respectively) for the Sonogashira coupling reactions (Scheme 2). The effects of catalysts were investigated using 4-nitrobromobenzene with different terminal acetylenes as shown in Table 2. To optimize the reactions, the reaction of 4-nitrobenzene and trimethylsilylacetylene was carried out at 30, 40 and 50 °C. Among these three reaction temperatures, 50 °C was found to be the best, as the reaction was found to be incomplete at other two temperatures (30 and 40 °C). The reaction temperature of 50 °C was, therefore, used for all the coupling reactions. It is to be noted that Sonogashira coupling reactions typically require reaction temperature of ~80 °C or even at higher temperature [33–35]. Using the new palladium(II) complexes as catalysts, it is possible to perform Sonogashira coupling reaction at a moderate temperature (50 °C) with good yields.

Sonogashira coupling reactions of 4-nitrobromobenzene with different terminal acetylenes using catalyst 1/catalyst 2
Using catalyst 1, the most efficient reaction (isolated yield: 90 %) was obtained from the reaction between 4-nitrobromobenzene and tolylacetylene (entry 4, Table 4). For catalyst 2, the highest yield was obtained in the case of phenylacetylene (entry 3, Table 4). Thus, our new palladium(II) complexes showed better catalytic efficiency in Sonogashira coupling reactions than the previously reported catalysts [33–35].
Similar catalytic activities using different palladium(II) complexes were reported in the literature. Hundertmark et al. [15] reported the catalytic activity of Pd(PhCN)2Cl2/P(t-Bu)3 in Sonogashira coupling reaction at room temperature. In our case, the palladium(II) complexes and their synthesis routes are different.
4 Conclusions
Palladium(II) nitroaryl complexes trans-[bromo(p-nitrophenyl)bis(triphenylphosphine)palladium(II)] 1 and trans-[bromo(2,4-dinitrophenyl)bis-(triphenylphosphine)palladium(II)] 2 have been synthesized by the oxidative addition of aryl bromide to tetrakis(triphenylphosphine)palladium(0), and characterized by FT-IR, NMR (1H, 13C and 31P) and elemental analysis. In complex 2, the square planar geometry of the palladium atom was confirmed by the single-crystal X-ray crystallography. Both the complexes showed better catalytic efficiency than typical catalysts used in the Sonogashira coupling reactions. The complexes exhibit intense luminescence peaks at 404 nm. Thermal studies showed that both complexes possess good thermal stability, and complex 1 is more stable than complex 2.
5 Supplementary Materials Available
Crystallographic data (comprising hydrogen atom coordinates, thermal parameters and full tables of bond lengths and angles) for the structural analysis has been deposited with the Cambridge Crystallographic Centre, CCDC No. 1478703 (2). Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (Fax: +44-1223-336-033; e-mail: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk).
References
M. Trivedi, G. Singh, R. Nagarajan, N.P. Rath, Inorg. Chim. Acta 394, 107 (2013)
B.N. Lin, S.H. Huang, W.Y. Wu, C.Y. Mou, F.Y. Tsai, Molecules 15, 9157 (2010)
J. Singh, A.K. Verma, J. Chem. Sci. 123, 937 (2011)
E.M. Beccalli, G. Broggini, M. Martinelli, S. Sottocornola, Chem. Rev. 107, 5318 (2007)
R.D. Stephens, C.E. Castro, J. Org. Chem. 28, 3313 (1963)
H.A. Dieck, F.R. Heck, J. Organomet. Chem. 93, 259 (1975)
L. Cassar, J. Organomet. Chem. 93, 253 (1975)
K. Sonogashira, Y. Tohda, N. Hagihara, Tetrahedron Lett. 16, 4467 (1975)
H. Doucet, J.C. Hierso, Angew. Chem. Int. Ed. Engl. 46, 834 (2007)
V. Polshettiwara, C. Len, A. Fihri, Coord. Chem. Rev. 253, 2599 (2009)
Y. Liang, Y.X. Xie, J.H. Li, J. Org. Chem. 71, 379 (2006)
B.J. Khairnar, S. Dey, V.K. Jain, B. M. Bhanage. Tetrahedron. Lett. 55, 716 (2014)
M. Islam, P. Mondal, K. Tuhinab, A.S. Roy, J. Braz. Chem. Soc. 22, 319 (2011)
K. Sonogashira, J. Organomet. Chem. 653, 46 (2002)
S.B. Park, H. Alper, Chem. Commun. 11, 1306 (2004)
T. Hundertmark, A.F. Littke, S.L. Buchwald, G.C. Fu, Org. Lett. 2, 1729 (2000)
D.D. Perrin, W.L.F. Armarego, Purification of Laboratory Chemicals (Pergamon Press, Oxford, 1988)
A.X.S. Bruker, SAINT, Reference Manual, Siemens Energy and Automation (Madison, 1994)
G.M. Sheldrick, G.M. SADABS, Empirical Absorption Correction Progra, (University of Göttingen, Göttingen, 1997)
G. M. Sheldrick, SHELXTL, Reference Manual, ver. 5.1, (Madison, 1997)
S. Komiya, Synthesis of Organometallic Compounds (Wiley, Chichester, 1997)
D. Taher, B. Walfort, H. Lang, Inorg. Chim. Acta 359, 1899 (2006)
S. Norio, A. Kimiyoshi, K. Takeo, Org. Lett. 6, 10 (2004)
H.P. Pradnya, A.F. Rodney, RSC Adv. 5, 54037 (2015)
B. Margherita, C. Silvano, D. Stefano, Eur. J. Org. Chem. 1 (2013)
X. Kai, S. Suyan, Z. Guodong, Y. Fan, W. Yangjie, RSC Adv. 4, 32643 (2014)
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds (Wiley, Hoboken, 1997)
H. Lang, D. Taher, B. Walfort, H. Pritzkow, J. Organomet. Chem. 691, 3834 (2006)
K.H. Yiha, G.H. Leeb, J. Chin. Chem. Soc. 55, 109 (2008)
P.M. Maitlis, The Organic Chemistry of Palladium (Academic Press, New York, 1971)
S.W. Lai, T.C. Cheung, M.C.W. Chan, K.K. Cheung, S.M. Peng, C.M. Che, Inorg. Chem. 39, 255 (2000)
Q.F. Zhi, L.Y. Xiao, F.Y. Yuan, Sci. World J. 1 (2013)
A.M. Asiri, S.A. Khan, Molecules 15, 4784 (2010)
X.D. Xu, J. Zhang, X. Yu, L.J. Chen, D.X. Wang, T. Yi, F. Li, H.B. Yang, Chem. Eur. J. 18, 16000 (2012)
I. Kim, A. Dunkhorst, J. Gilbert, U.H.F. Bunz, Macromolecules 38, 4561 (2005)
Acknowledgments
The financial support from the Higher Education Quality Enhancement Project (HEQEP) of the University Grants Commission (UGC) of Bangladesh is gratefully acknowledged. W.-Y.Wong acknowledges the financial support from the National Natural Science Foundation of China (Grant No.: 51373145), Hong Kong Research Grants Council (HKBU203312), Areas of Excellence Scheme, University Grants Committee of HKSAR (AoE/P-03/08), Science, Technology and Innovation Committee of Shenzhen Municipality (JCYJ 20140419130507116) and Hong Kong Baptist University (FRG2/13-14/083).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to the Special Issue in memory of Prof. Anatoli D. Pomogailo.
Rights and permissions
About this article

Cite this article
Huq, M.M., Robiur Rahman, M., Naher, M. et al. Synthesis, Characterization and Catalytic Activities of Palladium(II) Nitroaryl Complexes. J Inorg Organomet Polym 26, 1243–1252 (2016). https://doi.org/10.1007/s10904-016-0434-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-016-0434-3