
Abstract
Presented here are two d10 metal–organic coordination polymers (CPs), [Zn(C4O4)0.5(μ 3-OH)] n (1) and [Cd(C4O4)0.5(OX)0.5(H2O)] n (2) (H2OX = oxalic acid) constructed from squaric acid (H2C4O4) tectons. Single-crystal X-ray diffraction studies indicated that both 1 and 2 show 3D structures. The two 3D CPs are assembled from 1D {[Zn(μ 3-OH)] + n } chains to 3D structure linked through C4O4 2− ligands for 1 and 2D {[(Cd1)2(C4O4)]2+} layers pillared by OX2− ligands for 2. The structure of compound 1 can be described as a trinodal (3,6,6)-connected net with the point symbol of {43}2{44·610·8}{45·610}2, whereas 2 possess a trinodal (4,5,6)-connected {42·84} {46·66·83} {48·62}2 topology. Furthermore, the fluorescent and thermal stabilities properties of these two compounds were investigated.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The design and construction of new functional coordination polymers (CPs) have been one of the pioneer fields in current crystal engineering due to their intriguing topological structures and potential applications in diverse areas (e.g. catalytic, optical, molecular recognition, gas storage/separation, sensors, drug delivery, etc.) [1–5]. At present, large numbers of novel CPs have been designed and synthesized, but the reasonable design and controllable synthesis, however, is difficult to control since the final structure is frequently subject to subtle variation of external parameters, such as solvent [6], temperature [7], pH [8], organic ligands [9], metal cations [10], etc. Among these factors, the organic ligands influence profoundly the specific structures of synthesized products, and to dictate their formation with potential applications [11].
In recent years, the symmetrical, planar and rigid squaric acid has attracted much attention in the preparation of functional polymeric frameworks. It features extensive π-electron delocalization over all the oxygen and carbon atoms, which tends to induce hydrogen-bonding, π–π interactions for the construction of extended frameworks. As well as it has versatile coordination modes, so that it is widely used as a µ 2 to µ 8 bridging ligand bonded with metal ions in various bridging modes (Scheme 1) to create numerous amazing metal–squarate frameworks, including 1-, 2-, and 3-D cube and cage-like frameworks [12–18]. Recently, a few investigative examples of squarate-bridged metal complexes have been reported. For instance, cobalt-base complexes, Co3(OH)2(C4O4)2·3H2O [19] and Co3(OH)2(C4O4)2 [20] have been observed, which show interesting magnetic properties. Our groups have concentrated on utilizing squaric acid ligands and d10 metal ions to constructing novel topological networks [21], as an extension of our work, herein, we report the synthesis, structural characterizations, and fluorescent properties of two new CPs, namely, [Zn(C4O4)0.5(μ 3-OH)] n (1) and [Cd(C4O4)0.5(OX)0.5(H2O)] n (2).

Coordination modes of squarate on the construction of coordination polymers
2 Experimental
2.1 Materials and General Methods
All solvents and reagents for syntheses were commercially available and used as received. The infrared spectra (400–4000 cm−1) were recorded as KBr pellets a FTIR Nexus spectrophotometer. Elemental analyses were performed on a Perkin-Elmer 2400 Series II analyzer. X-ray powder diffraction (XRPD) patterns for microcrystalline samples were taken on a Rigaku Ultima IV diffractometer (Cu Kα radiation, λ = 1.5406 Å), with a scan speed of 8° min−1 and a step size of 0.02 deg in 2θ. The calculated XPRD patterns were simulated by using the single-crystal X-ray diffraction data. Thermogravimetric (TGA) curves were recorded on a NETZSCH STA 449C microanalyzer in air atmosphere at a heating rate of 10 °C min−1. Fluorescence spectra were recorded on a Hitachi F-4500 fluorescence spectrophotometer at room temperature.
2.2 Preparation of [Zn(C4O4)0.5(μ 3-OH)] n (1)
A mixture of H2C4O4 (11.4 mg, 0.10 mmol), Zn(ClO4)2·6H2O (37.2 mg, 0.10 mmol), and H2O (6 mL) was stirred at room temperature for 0.5 h. The reaction mixture was transferred to a 25 mL stainless steel reactor with Teflon liner and heated at 140 °C for 72 h. The reaction system was cooled to room temperature and colorless block crystals of 1 were obtained; yield, 67 % (9.23 mg) based on Zn. Anal. Calcd. (%) for C2HO3Zn: C, 17.35; O, 34.67; H, 0.73; Found: C, 17.29; O, 34.65; H, 0.78. IR (KBr pellet, cm−1): 3413 (s), 1596 (m), 1560 (m), 1411 (w), 1130 (w), 1095 (s), 848 (w), 799 (w), 714 (w).
2.3 Preparation of [Cd(C4O4)0.5(OX)0.5(H2O)] n (2)
A mixture of H2C4O4 (11.4 mg, 0.10 mmol), Cd(ClO4)2·6H2O (41.9 mg, 0.10 mmol), H2OX (12.61 mg, 0.1 mmol), and H2O (6 mL) was stirred at room temperature for 0.5 h. The solution was transferred to a 25 mL stainless steel reactor with Teflon liner and heated at 140 °C for 72 h. The reaction system was cooled to room temperature and colorless block crystals of 2 were obtained with a yield of 73 % (16.83 mg) based on Cd. Anal. Calcd. (%) for C3H2CdO5: C, 15.64; O, 34.71; H, 0.87; Found: C, 15.59; O, 34.77; H, 0.91. IR (KBr pellet, cm−1): 3485 (s), 3256 (m), 1617 (m), 14 92 (s), 1311 (s), 133 (s), 1080 (s), 706 (m), 634 (w).
2.4 X-ray Crystallography
Single-crystal X-ray diffraction analysis of 1 and 2 were carried out on a Rigaku XtaLAB mini diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) by using φ/ω scan technique at 296(2) K. The collected data were reduced using the program CrystalClear [22] and an empirical absorption correction was applied. The structures were solved by direct methods and refined based on F 2 by the full matrix least-squares methods using SHELXTL [23]. The H-atoms were assigned with common isotropic displacement factors and included in the final refinement by use of geometrical restrains. Generally, the positions of the C-bound H-atoms were generated by a riding model on idealized geometries. The crystal qualities of these two compounds were not good enough as indicated by the high R int value [24, 25]. The crystallographic data and selected bond lengths and angles for 1 and 2 are listed in Tables 1 and 2.
3 Results and Discussion
3.1 Crystal Structures Analysis of 1 and 2
Single-crystal X-ray analysis reveals that compounds 1 and 2 both crystallize in the monoclinic system, but in different space group; P2 1 /c for 1 and P2 1 /n for 2. The asymmetric unit of 1 contains one independent Zn(II) ion, one OH− anion, half a C4O4 2− anion, while the asymmetric unit of 2 contains one Cd(II) ion, half an OX2− anion, half a C4O4 2− anion and one coordinated water molecule. In 1, the Zn(II) center shows a distorted octahedral [ZnO6] sphere composed of six oxygen atoms from three hydroxyls and three C4O4 2− ligands (Fig. 1). The Zn–O bond lengths are in the range of 2.024(3)–2.543(3) Å, and the bond angles O–Zn–O are vary from 80.14(9)° to 168.28(10)° (Table 2). It should be mentioned that the Zn(1)–O(1)x+1/2, −y+3/2, z+1/2 (2.543(3) Å) bond distance is slightly longer than normal Zn–O bond distances. But it may be considered normal, when compared with the values of 2.550–2.711 Å cited in the literatures [26]. And in general, the long Zn–O distance indicates a relatively weak interaction, which can be considered to be secondary coordination. While for 2, Cd1 is eight-coordinated by four oxygen atoms from three different C4O4 2− anions, three oxygen atoms from two different OX2− anions, and one oxygen atom from water molecule (Fig. 2). The disposition of the eight donor atoms can be adequately described as a distorted dodecahedral geometry (Fig. S1). The Cd–O bond distances vary from 2.272(3) to 2.652(6) Å (Table 2). Observed Cd-donor atom bonds are in good agreement with values reported previously for eight-coordinated cadmium complexes [27, 28].

Coordination environment of the Zn center in compound 1. Symmetry codes for the generated atoms: #1: −x − 1, −y + 1, −z; #2: −x, −y + 1, −z; #3: x − 1, −y + 1/2, z − 1/2; #4: x − 1, y, z; #5: −x + 1, −y + 1, −z + 1 (Color figure online)

Coordination environment of the Cd center in compound 2. Symmetry codes for the generated atoms: #1: −x, −y + 2, −z + 1; #2: −x + 1, −y + 2, −z + 1; #3: x − 1, y, z; #4: x + 1/2, −y + 3/2, z + 1/2; #5: −x + 1/2, y + 1/2, −z + 3/2 (Color figure online)
Notably in 1, the hydroxyl adopts μ 3-bridging coordination mode to link the adjacent Zn(II) ions to form 1D {[Zn1(μ 3-OH)] + n } chain along the a axis (Fig. 3a). Furthermore, through sharing Zn(II) centers, the C4O4 2− anions in a μ 6-η 1 :η 2 :η 1 :η 2 coordination mode (Scheme 1p) connect these 1D chains to produce a 3D network (Fig. 3b). Besides, in 1, there exists strong hydrogen-bonding interactions from μ 3-OH group (O3) to O2 from C4O4 2−, (O3···O2 = 2.880 Å, O3–H···O2 = 162°). These weak interactions may make a certain contribution to the additional stability of the whole structure. From a topological point of view, each C4O4 2− anion can be viewed as a 6-connected node, each Zn(II) as 6-connected node, and each μ 3-OH as 3-connected node, so the whole framework can be defined as a rare trinodal (3,6,6)-connected net with the point symbol of {43}2{44·610·8}{45·610}2 (Fig. 3c).

a The 1D {[Zn(μ 3-OH)] + n } chains along c axis; b view of the 3D structure along the a axis. c Perspective view of the trinodal (3,6,6)-connected network of 1. Color codes: blue for 6-connected Zn(II), pink for 6-connected C4O4 2− ligand and red for 3-connected μ 3-OH (Color figure online)
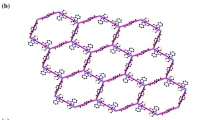
While in 2, the C4O4 2− anions with μ 6-η 2 :η 2 :η 2 :η 2 bridging coordination mode (Scheme 1o) link Cd(II) centers into a 2D {[(Cd1)2(C4O4)]2+} layer, which contains two different chains (Fig. 4a). Further examination shows that the two kinds of chains are consisted of four-member rings (purple in Fig. 4a) and ten-member rings (green in Fig. 4a), respectively. And the chains are associated with each other via edge-sharing the Cd–O bonds and result in formation of a 2D layer. Furthermore, the small molecule OX2− with μ 4-η 1 :η 2 :η 1 :η 2 coordination mode serves as pillar to join the neighboring 2D layers to a 3D framework (Fig. 4b). Topological analysis shows that this 3D network can be simplified by considering C4O4 2− as 6-connected node, OX2− as 4-connected node, and Cd(II) as 5-connected node, so the whole framework can be simplified as a trinodal (4,5,6)-connected network with the point symbol of {42·84} {46·66·83} {48·62}2 (Fig. 5).

a View of the 2D layer along a axis. Purple 1D chain consists of four-member rings. Green 1D chain consists of ten-member rings; b view of the 3D structure of 2. Hydrogen atoms and water molecules have been omitted for clarity (Color figure online)

Schematic representation of the trinodal (4,5,6)-connected network of 2. Color codes: blue for 5-connected Cd(II), golden for 6-connected C4O4 2− ligand and purple for 4-connected OX2− ligand (Color figure online)
It can be clearly seen that the structures of compounds 1 and 2 are different, although they were synthesized by the similar hydrothermal conditions except that different small molecule ligands (OH− or C2O4 2−) as well as different d10 metal ions [Zn(II) or Cd(II)] are employed. As mentioned above, the structure of compound 1 assembled from 1D {[Zn(μ 3-OH)] + n } chains to 3D structure linked through C4O4 2− ligands for 1, but 2D {[(Cd1)2(C4O4)]2+} layers to 3D network pillared by OX2− ligands for 2. The structural difference could mainly arise from the different coordinated modes of the C4O4 2− ligand (Scheme 1p for 1 and Scheme 1o for 2).
3.2 TGA and X-ray Powder Diffraction (XRPD)
The experimental and computer-simulated XRPDs confirm the purity of 1 and 2 (Fig. S2). The thermal stability of 1 and 2 were performed on single-phase polycrystalline samples from 20 to 800 °C. The TGA curve (Fig. S3) shows that 1 is thermally stable up to 370 °C, and thereafter significant weight loss occurs resulting in complete decomposition of the compound. The final residual weight is likely attributed to ZnO (calc. 58.8 % and exp. 58.1 %). While for 2, the TGA demonstrates that 2 shows a two-step weight loss and the sample is thermally stable up to170 °C. The first weight loss is 6.7 % from 170 to 210 °C and corresponds to the loss of coordination water molecules (calcd. 7.8 %). And the anhydrous composition is stable up to 320 °C. After 320 °C, the framework of 2 rapid collapses until 480 °C, which is attributed to the decomposition of 2. The total mass loss of 43.9 % corresponds to the removal of the organic species (calc. 44.3 %). The remaining weight (56.1 %) corresponds to CdO.
3.3 Fluorescent Property
Taking into account the excellent luminescent properties of d10 metal complexes, the solid-state luminescence of compounds 1 and 2 at room temperature was investigated (Fig. 6). It can be observed that the fluorescence emission occurs at around 396 nm for 1, under λ ex = 247 nm, whereas no detectable emission occurs for 2 under λ ex = 247 nm. To understand the nature of the emission band, the photoluminescence property of the squaric acid ligand was also analyzed. It found that two emission bands at 284 and 396 nm could be observed for free squaric acid ligand under λ ex = 240 nm. For 1, the complete quenching of the emission peak at 286 nm is probably attributed to the hydroxyl O–H oscillators [29]. And compound 2 does not exhibit detectable emission probably due to the quenching effects of the aqua ligands [30, 31].

Solid-state emission spectra of squaric acid, 1 and 2 at room temperature (Color figure online)
4 Conclusions
In summary, two new 3D polymeric Zn(II)/Cd(II)-networks have been successfully isolated under hydrothermal condition by reaction of squaric acid and different d10 metal salts. The different topological nets of compounds 1 and 2 are mainly caused by the different coordination modes of squarate. In addition, compared to squaric acid ligand, compound 1 and 2 have obvious fluorescence quenching due to the O–H vibrational excitation from the u-OH and the aqua ligand. Further investigations for the influence of d10 metal ions on squarate-based coordination frameworks are ongoing in our lab.
5 Supplementary Materials
CCDC 1010496 and 1010497 for 1 and 2, respectively, contain the supplementary crystallographic data for this paper. These CIF data can also be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridges CB2 1EZ, UK; fax: (+ 44) 1223-336-033; or email: deposit@ccdc.cam.ac.uk. Addition structural figures, TG and powder X-ray patterns can be found in the supporting file. Supplementary data associated with this article can be found, in the online version.
References
Y. Yue, Z.A. Qiao, P.F. Fulvio, A.J. Binder, C. Tian, J. Chen, S. Dai, J. Am. Chem. Soc. 135, 9572 (2013)
D.N. Bowman, J.H. Blew, T. Tsuchiya, E. Jakubikova, Inorg. Chem. 52, 8621 (2013)
D.S. Li, Y.P. Wu, J. Zhao, J. Zhang, J.Y. Lu, Coord. Chem. Rev. 261, 1 (2014)
T. Li, F.L. Wang, S.T. Wu, X.H. Huang, S.M. Chen, C.C. Huang, Cryst. Growth Des. 8, 3271 (2013)
D.S. Li, J. Zhao, Y.P. Wu, B. Liu, L. Bai, K. Zou, M. Du, Inorg. Chem. 52, 8091 (2013)
W.W. Dong, D.S. Li, J. Zhao, L.F. Ma, Y.P. Wu, Y.P. Duan, CrystEngComm. 15, 5412 (2013)
S. Chen, R.Q. Fan, C.F. Sun, P. Wang, Y.L. Yang, Q. Su, Y. Mu, Cryst. Growth Des. 12, 1337 (2012)
Y.P. He, Y.X. Tan, J. Zhang, Inorg. Chem. 52, 12758 (2013)
T. Li, M.T. Kozlowski, E.A. Doud, M.N. Blakely, N.L. Rosi, J. Am. Chem. Soc. 135, 11688 (2013)
T. Li, X. Liu, Z.P. Huang, Q. Lin, C.L. Lin, Q.G. Zhan, X.D. Xu, Y.P. Cai, Inorg. Chem. Commun. 39, 70 (2014)
C.C. Wang, C.H. Yang, G.H. Lee, Eur. J. Inorg. Chem. 2006, 820 (2006)
C.C. Wang, W.C. Chung, H.W. Lin, S.C. Dai, J.S. Shiu, G.H. Lee, H.S. Sheu, W. Lee, CrystEngComm. 13, 2130 (2011)
H. Erer, O.Z. Yesilel, N. Dege, Y.B. Alpaslan, J. Inorg. Organomet. Polym. Mater. 20, 411 (2010)
H.Y. Zang, H.N. Miras, J. Yan, D.L. Long, L. Cromin, J. Am. Chem. Soc. 124, 11376 (2012)
C.C. Wang, C.T. Kuo, J.C. Yang, G.H. Lee, W.J. Shih, H.S. Sheu, Cryst. Growth Des. 7, 1476 (2007)
P. Millett, L. Sabadie, J. Galy, J.C. Trombe, J. Solid State Chem. 173, 49 (2003)
A. Bouayad, J.C. Trombe, A. Gleizes, Inorg. Chim. Acta 230, 1 (1995)
S.L. Georgopoulos, R. Diniz, B.L. Rodrigues, L.F.C.D. Oliveira, J. Mol. Struct. 741, 61 (2005)
R.A. Mole, J.A. Stride, P.F. Henry, M. Hoelzel, A. Senyshyn, A. Alberola, C.J.G. Garcia, P.R. Raithby, P.T. Wood, Inorg. Chem. 50, 2246 (2011)
R.A. Mole, M.A. Nadeem, J.A. Stride, V.K. Peterson, P.T. Wood, Inorg. Chem. 52, 13462 (2013)
X.J. Ke, D.S. Li, J. Zhao, L. Bai, J.J Yang, Y.P. Duan. Inorg. Chem. Commun. 21, 129 (2012)
Rigaku, CrystalClear. Rigaku Americas, The Woodlands, Texas, USA, and Rigaku Corporation, Tokyo, Japan (2011)
G.M. Sheldrick, Acta Crystallogr. A64, 112 (2008)
X.H. Be, M.L. Rong, H.C. Chang, S. Kitagawa, S.R. Batten, Angew. Chem. Int. Ed. 43, 192 (2004)
L. Cunha-Silva, R. Ahmad, M.J. Carr, A. Franken, J.D. Kennedy, M.J. Hardie, Cryst. Growth Des. 7, 658 (2007)
A.A. Khandar, J. White, T. Taghvaee-Yazdeli, S.A. Hosseini-Yazdi, P. Mcardle, Inorg. Chim. Acta 400, 203 (2013)
X.Z. Wang, D.R. Zhu, Y. Xu, J. Yang, X. Shen, J. Zhou, N. Fei, X.K. Ke, L.M. Peng, Cryst. Growth Des. 10, 887 (2010)
A. Jabłońska-Wawrzycka, K. Stadnicka, J. Masternak, M. Zienkiewicz, J. Mol. Struct. 1012, 97 (2012)
H.B. Xu, J. Li, L.Y. Zhang, X. Huang, B. Li, Z.N. Chen, Cryst. Growth Des. 10, 4101 (2010)
Y.L. Gai, F.L. Jiang, K.C. Xiong, L. Chen, D.Q. Yuan, L.J. Zhang, K. Zhou, M.C. Hong, Cryst. Growth Des. 12, 2079 (2012)
M.D. Allendorf, C.A. Bauer, R.K. Bhakta, R.J. Houk, Chem. Soc. Rev. 38, 1330 (2009)
Acknowledgments
This work was financially supported by the NSF of China (Nos.: 21301106 and 21201109), the NSF of Hubei Province of China (2011CDA118), the Project of Hubei Provincial Education Office (Q20131304).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tang, P., Dong, WW., Xia, W. et al. Two New Zn(II)/Cd(II) Coordination Polymers Based on Rigid Squaric Acid: Crystal Structure, Topology and Fluorescent Properties. J Inorg Organomet Polym 25, 569–575 (2015). https://doi.org/10.1007/s10904-014-0118-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-014-0118-9