
Abstract
Employing the Molecular Electron Density Theory, [3 + 2] cycloaddition processes between 4-chlorobenzenenitrileoxide and linalool, have been applied using the DFT/B3LYP/6–311(d,p) method, activation, reaction energies and the reactivity indices are calculated. In an investigation of conceptual DFT indices, LIL-1 will contribute to this reaction as a nucleophile, whilst NOX-2 will participate as an electrophile. This cyclization is regio, chemo and stereospecific, as demonstrated by the reaction and activation energies, in clear agreement with the experiment's results, in addition, ELF analysis revealed that the mechanism for this cycloaddition occurs in two steps. Furthermore, a docking study was conducted on the products studied, and the interaction with the protein protease COVID-19 (PDB ID: 6LU7), our results indicate that the presence of the –OH group increases the affinity of these products, moreover, adsorption study by chromatography was made on silica gel as support; our outcome reveals that the –OH group creates an intramolecular hydrogen bond in the product P2, while in the product P3 will create a hydrogen bond with the silica gel which makes the two products P2 and P3 are very easy to separate by chromatography, this result is in excellent agreement with the Rf retention value. The study might provide a fundamental for developing natural anti-viral compound in promoting human health.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Heterocyclic synthesis is a field of chemistry that involves the production of cyclic molecules that include atoms other than carbon in the ring [1,2,3,4]. Heterocyclics are frequently used in many chemicals, including drugs, pigments, and flavors, making them important for many industrial and scientific applications [5,6,7,8]. There are several methods for synthesizing heterocyclic, the most interesting of which is intermolecular cyclization [9,10,11], as an example 1,3-dipolar cycloaddition reaction, in this a chemical reaction in which two molecules, add together to form a ring [12,13,14]. This reaction is characterized by the formation of a covalent bond between two distant atoms in the reactants, resulting in the formation of a new ring. 1,3-dipolar cycloaddition reactions are very useful for the synthesis of complex molecules like isoxazolines [15,16,17].
The isoxazolines includes a group of organic compounds that have in common the presence of an isoxazoline ring in their chemical structure (Fig. 1). Isoxazolines can be produced by different methods, such as oxazole-nation reaction or condensation of carbaldehydes with amines and 1,3-dipolar cycloaddition reaction [18,19,20,21]. Isoxazolines are used in many industrial applications, such as herbicides, insecticides, and drugs. Due to their unique properties, such as stability, and biological activity, isoxazolines are becoming increasingly popular in organic synthesis and to improve the biological activity of isoxazolines [22,23,24,25], their group is replaced by monoterpenes such as linalool.

Structures of isoxazolines and isoxazole
Linalool is a colorless mono-terpene alcohol molecule, produced by several plants including lavender, basil, mint, and coriander, and is widely used in perfumery; linalool has also been studied for its potential medicinal properties and has been found to have analgesic, anxiolytic and anti-inflammatory effects [2], There is a lot of potential for linalool to be used as a secure and natural therapeutic substitute. Many studies concentrate on the bioactive properties of linalool, including their modes of action and their anticancer, antibacterial, neuroprotective, anxiolytic, antidepressant, anti-stress, hepatoprotective, kidney protective, and lung protective activity. Additionally, the possibility of encapsulating linalool and the medicinal potential of linalool are examined. Through oxidative stress, linalool can cause cancer cells to die while simultaneously protecting healthy cells. Cell membrane disruption is how linalool exerts its antibacterial properties. The anti-inflammatory properties of linalool are what cause it to have beneficial effects on the liver, kidneys, and lungs. Linalool can be utilized as an adjuvant to antibiotics or anti-cancer medications due to its protective properties and minimal toxicity [2]. To increase the properties of this molecule, several reactions have been made as example the 1,3-dipolar cycloaddition reaction, the dipole used is 4-chlorobenzenenitrileoxide which leads to two products. (Fig. 2). (Rf formula is given supporting information (Fig. S1).

Cycloaddition reaction between 4-ChlorobenzeneNitrileOxide and linalool (Rf: retention value. (Rf formula is given supporting information (Fig. S1))
The molecular electron density theory (MEDT) ability to consistently that the molecular reactivity is controlled by an electron density, in the context, numerous chemical reactions, including the [3+2] [26,27,28,29,30,31,32,33,34,35] and [4+2] cycloadditions [36, 37], the epoxidation reaction [38,39,40,41], and the nitration reaction [42], have been explored, also, we have revealed that the MEDT theory offers more details than the FMO theory (Frontier Molecular Orbital Theory) [43].
Within the framework of the theory (MEDT), we used the most educated and reliable method Density functional theory DFT/B3LYP/6-311(d,p) [38,39,40,41,42,43,44,45,46], to explain the cycloaddition reaction of Linallol and Chlorobenzene-nitrile-oxide experimentally explored by Rouani et al [44] and its mechanism, chemoselectivity, regioselectivity, and stereoselectivity (Fig. 2), moreover, we performed out a docking evaluation for the potential products of this reaction (Fig. 3) and we also performed a silica gel adsorption study of the two products P2 and P3 to understand why the two products are very easy to separate by chromotography on silica gel.

a docking evaluation for the potential products
Quantum Calculation Methods
All the computations are processed in Gaussian 09 [47] utilizing DFT/B3LYP/6–311(d,p) [44, 45] and GaussView5 to visualize reagents and products. The IRC (Intrinsic Reaction Coordinate) has been utilized to survey the reaction path from its initial state to its final state and the influence of the reaction solvent is counted by the Tomasi model (PCM). (Polarizable Continuum Model) [48,49,50,51,52,53]. The electronic chemical potential is designated:\({\varvec{\mu}}=\frac{{E}_{L}+{E}_{H}}{2}\). The Chemical hardness is defined by:\(\eta ={E}_{L}-{E}_{H}\), using the energies of the HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) boundary molecular orbitals, are given by EH and EL. The global electrophilicity index (w) is a quantity used in chemistry to measure the tendency of a molecule to react as an electrophile is defined by the following formula: \(\upomega =\frac{{\mu }^{2}}{2\eta }\) [54], contrary to the index of electrophilicity, the index of nucleophilie (N) is a concept useful to check the tendency of a molecule to react as a nucleophile, i.e. to give electrons to another molecule to form a chemical bond, defined as: E = EHOMO (Nu)-EHOMO (TCE) [55]. Parr functions have been employed to find local indices, which are basic spin wave functions that are used in quantum chemistry to describe the electronic spin distribution in molecules [56, 57]. The electron localization function was described employing a Topmod software (ELF), this ELF is used to provide a representation of the distribution of electrons and electron cavities in the reagents [58, 59].
Adsorption Analysis Using Mds
MDs are an especially useful method to inspect the exact configuration for absorbates (two organic molecules)-substrate (silica surface) systems and determine the most stable adsorption configurations [60]. In this current research work, the Molecular dynamics computer simulations were executed in BIOVIA Materials Studio®Dassault Systèmes (formerly Accelrys) software (2020 v20.1.0.2728) [61]. At the beginning of two P2&P3 molecules, in the gas phase. Because the COMPASS force field takes into account different types of interactions, such as covalent bonds, electrostatic interactions, van der Waals interactions, etc. moreover, the positions and velocities of atoms are tracked over time to understand the dynamic behavior of a system. It is based on empirical parameters derived from experimental and theoretical data in order to best reproduce the properties and molecular behaviors observed the reason why the geometrically optimized by utilizing COMPASS force field in Forcite module. The purpose of these Molecular dynamics computer simulations is to look at how the two products P2 and P3 interact with a Silicon dioxide surface in the gas phas. Thereafter geometry optimization by BIOVIA Materials Studio®Forcite model of the constructed solution slab was performed. Since the Silicon dioxide (SiO2(110)) surface(Silica gel surface) has a tightly packed structure and is more energetically stable than other planes, it was chosen for this investigation instead of Silicon dioxide (SiO2(110)). After that, the optimized constructed solution slab is placed above the (SiO2(110)) surface maintaining a 10 Å vacuum slab as blank space at the uppermost layer to obtain a simulation model of (54,052*85,095*39,119Å). Before running MDs, the geometry of the built-in simulation model was tuned to prevent undesirable molecule configurations and produce an energy-optimal simulation setup. The stabilization of the energy and temperature fluctuation curves proved that the optimization approach had been effective. The Forcite module in BIOVIA Materials Studio®-Dassault Systems was employed to perform the Molecular dynamics computer simulations utilizing the COMPASS force field and NVT ensemble with a time step of (1 fs) for (100 ps) at 298 K. The MDs results showed that the targeted two products P2 and P3 become spontaneously interacting with (SiO2(110)) surface atoms after the was completed. The following equation was used to calculate the interaction energy (Einteraction) of P2 and P3 with (SiO2(110)) surface [60].
Whereas Etotal represents the total energy for adsorbate–substrate,
Esurface represents the energy surface of (SiO2(110)) containing the adsorbate (Simple organic molecules (two products P2 and P3) and Eadsorbate represents the energy of the adsorbed. Again, the binding energy (Ebinding) is the negative value of Einteraction and it is determined as follows:
Results and Discussion
Computation of the CDFT Indices for the Reagents
Evaluating the reactivity indices providing inside CDFT is an effective procedure for understanding the reactivity of organic compounds, as revealed by numerous studies on chemical reactions [26,27,28,29,30,31]. Electrophilicity, nucleophilicity, chemical hardness, and electronic chemical potential, which are the global indices listed in Table 1, are explored in order to predict the reactivity of Linollol (LIL-1) and nitriloxide (NOX-2) in the cycloadition reaction.
Table 1 demonstrates that the electronic chemical potential of linalool (LIL-1) (μ = -2.88 eV) is minor, in absolute value, then that of nitriloxide (NOX-2) (μ = -4.04 eV), the global electron density transfer (GEDT) will proceed from LIL 1 to NOX-2.
Linalool (LIL-1) electrophilicity and nucleophilicity indices are calculated as w = 0.65 eV and N = 3.47 eV, respectively. As a result, this substance can be categorized as being both a strong nucleophile and a moderate electrophile. Since NOX-2 has an N nucleophilicity of 3.06 eV and an w electrophilicity of 1.68 eV, it is classifiable as a strong nucleophile and a strong electrophile. Clearly, NOX-2 will involve in this reaction as an electrophile and LIL-1 will engage in this reaction as a nucleophile.
Due to the similar properties of both reactants, we shall categorize them using transition state charge transfers, from LIL-1 to NOX-2 transition state charge transfer will take place, the fact that LIL-1 will act as a nucleophile and NOX-2 will behave as an electrophile.
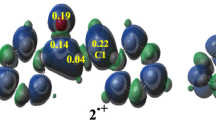
Because this 32CA reaction is non-polar, many studies show that the formation of the first bond in cycloaddition reactions takes place on the more electrophilic center of ethylene derivatives [26]. The electrophilic Parr functions of LIL-1 have been represented in Fig. 4.

B3LYP/6–311(d,p) Parr functions \({P}_{k}^{+}\) of Linalool 1
Figure 4 shows that the Mulliken atomic spin densities P+ of linalool are localized on the double bond C1 = C2 (0.33; 0.21); and almost less area around the double bond C5 = C6 (0.17;0.17). This result showed that the double bond C1 = C2 is the most reactive part of linalool, which confirms the full chemoselectivity observed in experiment.
ELF Study of the Reagents
Electron location function (ELF) is important in chemistry because they determine the physical and chemical characteristics of atoms and molecules, such as their reactivity, polarity and molecular geometry. They are employed to explain the formation of chemical bonds, the reactivity of molecules and the interactions between molecules in chemical reactions, in order to identify the electrical structure of the reactants, (Linalool and Nitrile oxide); a topological ELF study of the two reactants was realized. The ELF localization domains and valence basin populations are arranged in Fig. 5.

The ELF localization domains and valence basin populations
ELF examination of the linalool reveals the existence of two bisynaptic basins linked to the C1-C2 and C6-C7 atoms with a value of V(C1, C2) = 3.53e and V(C6,C7) = 3.80e respectively, two other bisybaptic basins of value V(O,C3) = 1.25e and V(C5,C6) = 1.98e.
Analysis of Energy
The two reactants are asymmetric and we have two doubles bond in linalool, so we will have eight cycloaddition reaction pathways, we will study five pathways, two pathways to prove the stereoselectivity and three others to explain the regio and chemioselectivity. Consequently we have located eight transition states named Ts-1, Ts-2, Ts-3, Ts-4 and Ts-5 the cyclic products corresponding to each transition state have been also characterized. The reactions activations relative energies are arranged in Fig. 6, Fig. 5 represents the profile of free energy and the different transition state geometries are given in Fig. 7, while the supplemental file contains the total calculation (Table S1).

Possible reaction routes in the cycloaddition process between linalool (LIL 1) and Chlorobenzenenitrileoxide (NOX 2) in ethanol

Enthalpy profile, for the reaction pathways studied of the cycloaddition reactions between linalool (LIL 1) and Chlorobenzenenitrileoxide (NOX 2) in ethanol
The transition states Ts-1, Ts-2, Ts-3, Ts-4, and Ts-5 have respective values of 17.69 kcal/mol, 9.95 kcal/mol, 14.70 kcal/mol, 21.05 kcal/mol, and 19.08 kcal/mol, designating that P1 product is kinetically more favorable. The exothermic characters of these products are: P1 (35.13kcal/mol), P2 (41.20 kcal/mol), P3 (40.23 kcal/mol), P4 (31.74 kcal/mol), and P5 (34.64 kcal/mol), It reveals that the item P2 is thermodynamically favorable, thus the product P2 is gained under kinetic and thermodynamic control.
Entropy is negative to the bimolecular process, causes the free Gibbs relative energies to increase intensely when the term TS is taken into account in comparison to the relative enthalpies of 13–14 kcal/mol, Fig. 5 illustrates that the free activation energy of the 2 + 3 cycloaddition reaction paths between linalool (LIL 1) and Chlorobenzenenitrileoxide (NOX 2) in ethanol are: Ts-1 (31.64 kcal/mol), Ts-2 (22.96 kcal/mol) Ts-3 (27.51 kcal/mol), Ts-4 (34.62 kcal/mol) and Ts-5 (33.35 kcal/mol), which indicates that pathway 2 is more favorable, so P2 is the favored product, consequently the 2 + 3 cycloaddition reaction between linalool (LIL-1) and Chlorobenzenenitrileoxide (NOX-2) is regiospecific, in addition, the energetic difference between Ts-2 and Ts4 is very great (11.66 kcal/mol), difference between Ts-2 and Ts3 is (4.55 kcal/mol) indicates that the 2 + 3 cycloaddition reaction between linalool (LIL-1) and chlorobenzenenitrileoxide (NOX-2) is stereo- and chemo-selective, this outcome is consistent with the experiment.
Figure 8 illustrates the geometries of the transition states corresponding to the five chemical pathways.

The different transition state geometries that were localized in the cycloaddition reaction between linalool (LIL 1) and chlorobenzenenitrileoxide (NOX 2)
The geometric parameters of the transition states shown in Fig. 5 allow for significant conclusions:
-
(i)
According to the bond lengths, evidently, the new single bonds are created in an asynchronous manner.
-
(ii)
The polar character of these cycloadditions reactions was evaluated by analyzing the values of the GEDT (Global Electron Density Transfer) at the corresponding TS at 0.03 TS-1, 0.04; at TS-2, 0.04; at TS-3, 0.04; at TS-4, and 0.03 at TS-5 these values clearly indicate that these cycloaddition reactions have a non-polar character.
BET Study
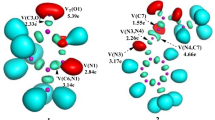
Bond evolution theory is a theory in chemistry that describes how chemical bonds between atoms are evolving during a chemical reaction. This theory was developed to provide an explanation of how molecules are formed and change in chemical reactions. According to this theory, chemical bonds evolve from transition states that form during a chemical reaction. This theory is widely used to understand the mechanisms of chemical reactions; in part evolutions of new bonds have been controlled and followed using Topmod software. Figure 9 illustrates the locations of the associated ELF basin attractors, while Table S2 displays information on the populations of the most important ELF valence basins that were contributing to the formation of C–C and C-O single bonds in the selected structures.

The locations of the corresponding ELF basin attractors
The C1 structure contains two monosynaptic basins carried by oxygen O1 of nitrileoxide of total value V(O1)=5.65e and two bisynaptic basins located between C1-N and N1-O1 of values respectively V(C1-N)= 5.93e and V(N-O1)= 1.64e, another bisynaptic basins located between C2-C3 of value V(C1-N)=3. 41e, which indicates this structure corresponding to the structures of the separate reactants, these two basins vary from structure C1 to structure C4, in structure C5 appearance of a monosynaptic basin carried by the carbon C1 of value V(C1)=0. 05e, this value increases and becomes 0.43e in structure C7 in the same structure we observe the appearance of two other monosynaptic basins carried by the oxygen O1 of value V(O1) = 0.11e and another carried by the carbon C2 of value V(C2)=0. 26e, these values become V(C1)=0.65e and V(C2) = 0.45e in the C9 structure, the two monosynaptic basins V(C1) and V(C2) are going to form the first C1-C2 bond at a distance of 2.002A, at this distance we have the formation of 61% of the C1-C2 bond.
In the C10 structure, we have the appearance of a monosynaptic basin carried by the C3 carbon of value V(C3)= 0.12e, this value becomes 0.16e in the C11 structure and the value of the basin V(O1) becomes 0.40e, the basins V(O1) and V(C3) are going to form the second single bond at a distance 1.753A at this distance 39% of the O1-C3 bond has been formed. This ELF study clearly shows that the cycloaddition reaction between Linallol and Chlorobenzene-nitrile-oxide followed a two-step mechanism.
Docking Survey
Coronavirus Disease-2019 (COVID-19) is a respiratory disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) which shares a close relation to RNA viruses [62,63,64,65,66]. Herein, theoretical approaches for the discovery of new antiviral drugs for COVID-19 were carried out using molecular docking studies with the SARS-CoV-2 main protease (PDB ID: 6LU7).
Molecular docking was performed to examine the antiviral properties of the title compound on the SARS-CoV-2 main protease protein. The P1-P5 ligands were converted to “PDB” using Gaussian 09 to study the protein–ligand interactions [47]. Autodock 4.2.6 program along with the graphical interface AutoDockTools (ADT) version 1.5.6 [67] has been used to analysis the binding modes of P1-P5 ligands with SARS-CoV-2 main protease (PDB code: 6LU7) which was collected from the RCSB Protein Data Bank (http://www.rcsb.org) to study the interactions of compounds with the active site of receptor. Before docking protocol, water molecules and the co-crystallized ligand were removed from the 6LU7 protein using Discovery Studio Software [68]. Polar hydrogen atoms were added to the crystal structures of the protein, followed by the addition of Kollman and Gasteiger charges atoms. Finally, pdbqt files of ligands and protein were generated. The grid box with radius of 60 × 60 × 60 Å3 in x, y and z directions was generated and the dimensions of the active site coordinates are -10.729 Å, 12.418 Å and 68.816 Å by the ligand location in the protein.
Molecular docking investigation of P1-P5 ligands was enhanced with bond lengths in Å units using the PyMol software [69]. Figure 10 depicts the 3D and 2D interaction diagrams between the target 6LU7 protein and the investigated P2-P5 ligands.

3D and 2D interaction diagrams of P1-P5 compounds with SARS-CoV-2 receptor PDB ID:6LU7
The Fig. 10 revealed that conventional hydrogen bonds formed between the H atom of the O–H group and GLU166, GLN189, GLU166, HIS163, and ARG188, respectively. The H atoms show the interactions with the GLU166, GLN189, GLU166, HIS163, and ARG188 with the distance of 8.6 Å, 1.9 Å, 4.6 Å, 1.8 Å, and 5.1 Å, respectively. From the 2D plot in Fig. 10, the P1, P4, P5 derivatives presented in blue color interact with ASP187 (5.4 Å, ASP187 (5.9 Å), ASP187 (3.6 Å) through the halogen (Cl, Br, I) bonds, respectively.
Details of the protein–ligand interaction patterns such as binding energy, inhibition constant and intermolecular energy of the P1-P5 derivatives with the 6LU7 targeted protein were depicted in Table 2. The binding energies with drug activeness against the targeted 6LU7 protein are found to be in the order of P5 > P4 > P3 > P2 > P1. The most active compound, P5, gave the highest binding energy (-7.83 kcal/mol), while the other compounds P1, P2, P3, and P4 created interaction energies of -7.33 kcal/mol, -7.40 kcal/mol, -7.55 kcal/mol, and -7.57 kcal/mol, respectively. The docking analysis of the P1-P5 derivatives with SARS-CoV-2 reveals that the compound P5 should show significant SARS-Cov-2 inhibitory activity.
We have revealed that the reaction between LIL and OX is highly regio, stere and chemoselective, we also tested the products formed against coronavirus, in the next section we will examine the adsorption of the products P2 and P3 on a silica gel to understand why these two products are easy to separate by chromatography.
Molecular Dynamic Simulation-Chromatography Study
Silica gel adsorption is a commonly employed process in chromatography to separate components of a mixture. Silica gel is a porous material, often in the form of small beads or powder, which has a large internal surface area. The components of a mixture can be separated using differences in their affinities for silica gel, which acts as the adsorbent in this case, in silica gel chromatography; the mixture to be separated is placed on the silica gel column. Molecular dynamic simulation MDs has developed as a cutting-edge approach for studying adsorbate–adsorbate interactions [70]. It also aids in visualizing the single (P2 and P3) molecules actual adsorption arrangement. The system's energy and temperature fluctuation curves provided evidence that the system had successfully attained equilibrium (Fig. 11).


The system's energy and temperature fluctuation curves for SiO2-P2 and SiO2-P3 during NVT dynamics run
It is evident that the P2 and P3 system's energy and temperature fluctuation curves converged to a persistent value with small oscillations at the end of the NVT MDs-runs; indicating that the both systems have reached the equilibrium condition. The most-stable adsorption configuration of two products P2 and P3 on the silicon dioxide surface (110) in the gas phase after NVT runs are illustrated in Fig. 12.

The configuration of P2 and P3 on SiO2(110) surface in horizontal configuration calculated by MDs. SiO2-P2 Complex: Eads = -18,252 kJ/mol
The Fig. 13 presented the resulting interaction (Einteraction) owing to the interaction of each organic molecules P2 & P3 with the surface of (SiO2(110)) in the gas phase. According to Molecular dynamics computer simulations results, the two products P2&P3 can interact with the (SiO2(110)) surface and subsequently adsorb on the (SiO2(110)) surface. The obtained interaction value of two products P2 & P3 suggests that P3 interacts more than P2 (Fig. 11). The calculated binding or interaction energy follows the order P3 > P2 (Fig. 13).

The resulting interaction value of two products P2 & P3 on SiO2(110) surface in horizontal configuration
Molecular dynamics computer simulations result revealed that the two products P2 and P3 are horizontally adsorbed on the (SiO2(110)) surface. The parallel adsorption of the of two products P2 and P3 is caused by the interaction of the heteroatoms present in the of two molecules P2 and P3 with surface (SiO2(110)). The horizontal fashion of adsorption of the two products P2 and P3 covers a greater surface area. Again, the adsorption configuration of two products P2 and P3 revealed that the both molecules are horizontally disposed over the surface (SiO2(110)). The horizontal adsorption orientation becomes feasible by the interaction of (SiO2(110)) surface and heteroatoms present in the P2 and P3 skeleton. Increased surface coverage on the (SiO2(110)) surface occurs from of two products P2 and P3 horizontal mode of adsorption. In comparison to of two products P2 and P3 adsorption becomes more feasible for P3. Now from the molecular point of view, P3 possesses one vacant site and by using that vacant cite of two products P2 and P3 may adsorb on the silicon dioxide substrate. The study of adsorption by MDs reveals that the –OH group creates an intramolecular hydrogen bond in the product P2, while in the product P3 will create a hydrogen –H bond with silica gel, which makes both the products P2 and P3 very easy to separate by chromatography. Both Einteraction values determined from MDs are well corroborated with the experimental findings.
Conclusion
The B3LYP/6–311(d,p) on density functional theory was used to analyze the cycloloaddition reaction [3 + 2] between linallol and chlorobenzene-nitrile-oxide. The analysis of reactivity indices, reaction energy, and activation shows that the cycloloaddition reaction follows five distinct paths. An exploration of conceptual DFT indices indicates that Linallol (LIL 1) will engage in this reaction as a nucleophile, whereas chlorobenzene-nitrile-oxide (NOX 2) will contribute so as an electrophile. The reaction and activation energies clearly show that this cyclization is regio- chemo and stereospecific, which is in perfect agreement with the outcomes of the experiment. The BET analysis reveals clearly that this reaction follows a two-step reaction mechanism. In addition adsorption study by MDs reveals that the –OH group creates an intramolecular hydrogen bond in the product P2, while in the product P3 will create a hydrogen –H bond with silica gel, which makes both the products P2 and P3 very easy to separate by chromatography. The inquiry into Mpro-COVID-19 suppression by pyrazoline compounds has produced promising findings. For the fight against the COVID-19 virus, it would be worthwhile to do more precise studies on this family of products.
Data Availability
No data was used for the research described in the article.
References
Franzén GR (2000) Recent Advances in the Preparation of Heterocycles on Solid Support: A Review of the Literature. J Comb Chem 2:195–214. https://doi.org/10.1021/cc000002f
An Q, Ren J-N, Li X, Fan G, Qu SS, Song Y, Lia Y, Pan SY (2021) Food Funct 12:10370–10389. https://doi.org/10.1039/D1FO02120F
Aparna EP, Devaky KS (2019) Advances in the Solid-Phase Synthesis of Pyrimidine Derivatives. ACS Comb Sci 21:35–68. https://doi.org/10.1021/acscombsci.8b00172
Alexander D, Stephen FM (2004) Synthesis of Oxygen- and Nitrogen-Containing Heterocycles by Ring-Closing Metathesis. Chem Rev 104(5):2199–2238. https://doi.org/10.1021/cr0200872
Alamgir ANM (2018) Secondary Metabolites: Secondary Metabolic Products Consisting of C and H; C, H, and O; N, S, and P Elements; and O/N Heterocycles. In: Therapeutic Use of Medicinal Plants and their Extracts. Prog Drug Res 2:74. https://doi.org/10.1007/978-3-319-92387-1_3
Ravinder K, Thakor R, Antonio M (1994) An overview of pigment production in biological systems: Functions, biosynthesis, and applications in food industry. Food Rev Intl 10(1):49–70. https://doi.org/10.1080/87559129409540985
Sagar NA, Pareek S, Benkeblia N, Xiao J (2022) Onion (Allium cepa L.) bioactives: Chemistry, pharmacotherapeutic functions, and industrial applications. Food Frontiers 3:380–412. https://doi.org/10.1002/fft2.135
Xia B, Ni ZJ, Hu LT, Elam E, Thakur K, Zhang JG, Wei ZJ (2021) We Development of meat flavors in peony seed-derived Maillard reaction products with the addition of chicken fat prepared under different conditions. Food Chem 363:130276. https://doi.org/10.1016/j.foodchem.2021.130276
Choury M, Basilio A, Blond G, Gulea M (2020) Synthesis of Medium-Sized Heterocycles by Transition-Metal-Catalyzed Intramolecular Cyclization. Molecules 25:3147. https://doi.org/10.3390/molecules25143147
Lv Y, Meng J, Li C, Wang X, Ye Y, Sun K (2021) Update on the Synthesis of N-Heterocycles via Cyclization of Hydrazones (2017–2021). Adv Synth Catal 363:5235. https://doi.org/10.1002/adsc.202101184
Wiebe A, Gieshoff T, Möhle S, Rodrigo E, Zirbes M, Waldvogel SR (2018) Electrifying Organic Synthesis. Angew Chem Int Ed 57:5594. https://doi.org/10.1002/anie.201711060
Payne K, White M, Fisher K (2015) New cofactor supports α, β-unsaturated acid decarboxylation via 1,3-dipolar cyc loaddition. Nature 522:497–501. https://doi.org/10.1038/nature14560
Lutz JF (2007) 1,3-Dipolar Cycloadditions of Azides and Alkynes: A Universal Ligation Tool in Polymer and Materials Science. Angew Chem Int Ed 46:1018–1025. https://doi.org/10.1002/anie.200604050
Kowalski K (2023) A brief survey on the application of metal-catalyzed azide–alkyne cycloaddition reactions to the synthesis of ferrocenyl-x-1,2,3-triazolyl-R (x = none or a linker and R = organic entity) compounds with anticancer activity. Coord Chem Rev 479:214996. https://doi.org/10.1016/j.ccr.2022.214996
Lauria A, Delisi R, Mingoia F, Terenzi A, Martorana A, Barone G, Almerico AM (2014) 1,2,3-Triazole in Heterocyclic Compounds, Endowed with Biological Activity, through 1,3-Dipolar Cycloadditions. Eur J Org Chem 3289–3306. https://doi.org/10.1002/ejoc.201301695
Satya KA, Saeed Ul, Sobia AH, Khan A, Muhammad U, Anwar R, Al-Harrasi A (2023) Synthesis of novel substituted quinoline derivatives as diabetics II inhibitors and along with their in-silico studies. J Mol Struct 1274(1):134560. https://doi.org/10.1016/j.molstruc.2022.134560
Jingli H, Xifang L, Shen J, Guilong Z, Wang PG (2012) The impact of click chemistry in medicinal chemistry. Expert Opin Drug Discov 7(6):489–501. https://doi.org/10.1517/17460441.2012.682725
Karola R-B, Freysoldt HE, Wierschem F (2005) 1,3-Dipolar cycloaddition on solid supports: nitrone approach towards isoxazolidines and isoxazolines and subsequent transformations. Chem Soc Rev 34:507–516. https://doi.org/10.1039/B311200B
Xiao-Wei Z, He X-L, Nan Y, Hong-Xing Z, Xiang-Guo Hu (2020) Oxidize Amines to Nitrile Oxides: One Type of Amine Oxidation and Its Application to Directly Construct Isoxazoles and Isoxazolines. J Org Chem 85(23):15726–15735. https://doi.org/10.1021/acs.joc.0c02281
Plumet J (2020) 1,3-Dipolar Cycloaddition Reactions of Nitrile Oxides under “Non-Conventional” Conditions: Green Solvents, Irradiation, and Continuous Flow. J Plumet, Chem Plus Chem 85:2252. https://doi.org/10.1002/cplu.202000448
Qiang F, Huang H, Jianwei S (2021) Ru-Catalyzed [3 + 2] Cycloaddition of Nitrile Oxides and Electron-Rich Alkynes with Reversed Regioselectivity. Org Lett 23:2431–2436. https://doi.org/10.1021/acs.orglett.1c00273
Egorova SK, Evgeniy Gordeev G, Ananikov PV (2017) Biological Activity of Ionic Liquids and Their Application in Pharmaceutics and Medicine. Chem Rev 117:7132–7189. https://doi.org/10.1021/acs.chemrev.6b00562
Jhonsi MA, Ananth DA, Nambirajan G, Sivasudha T, Yamini R, Bera S, Kathiravan A (2018) A Antimicrobial activity, cytotoxicity and DNA binding studies of carbon dots. Spectrochim Acta Part A Mol Biomol Spectrosc 196:295–302. https://doi.org/10.1016/j.saa.2018.02.030
Chakraborty B (2019) Mechanochemical synthesis and cycloaddition reactions of fluoro nitrone under solvent-free conditions and potential antimicrobial activities of the cycloadducts. J Heterocyclic Chem 56:3414–3422. https://doi.org/10.1002/jhet.3736
Angelika S, Obmińska-Mrukowicz B (2017) Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur J Med Chem 137:292–309. https://doi.org/10.1016/j.ejmech.2017.06.002
Ouahdi Z, Ourhriss N, El Idrissi M et al (2022) Exploration of the mechanism, chemospecificity, regiospecificity and stereoselectivity of the cycloaddition reaction between 9α-hydroxyparthenolide and nitrilimine: MEDT study. Theor Chem Acc 141:50. https://doi.org/10.1007/s00214-022-02913-6
El Idrissi M, El Ghozlani M, Eşme A, Ríos-Gutiérrez M, Ouled Aitouna A, Salah M, El Alaoui El Abdallaoui H, Zeroual A, Mazoir N, Domingo LR (2021) Mpro-SARS-CoV-2 Inhibitors and Various Chemical Reactivity of 1-Bromo- and 1-Chloro-4-vinylbenzene in [3 + 2] Cycloaddition Reactions. Organics 2:1–16. https://doi.org/10.3390/org2010001
Zeroual A, Ríos-Gutiérrez M, El Ghozlani M, El Idrissi M, Ouled Aitouna A, Salah M, El Alaoui El Abdallaoui H, Domingo LR (2020) A molecular electron density theory investigation of the molecular mechanism, regioselectivity, stereoselectivity and chemoselectivity of cycloaddition reaction between acetonitrile N-oxide and 2,5-dimethyl-2H-[1,2,3]diazarsole. Theoret Chem Acc 139:37. https://doi.org/10.1007/s12039-019-1656-z
Zeroual A, Ríos-Gutiérrez M, Salah M, El Abdallaoui El Abdallaoui H, Domingo LR (2019) An investigation of the molecular mechanism, chemioselectivity and regioselectivity of cycloaddition reaction between acetonitrile N-Oxide and 2,5-dimethyl-2H-[1,2,3]diazaphosphole: a MEDT study. J Chem Sci 131:75
El idrissi M, Eşme A, Hakmaoui Y, Ríos-Gutiérrez M, Ouled Aitouna A, Salah M, Zeroual A, Domingo LR (2021) Divulging the Various Chemical Reactivity of Trifluoromethyl-4-vinyl-benzene as well as Methyl-4-vinyl-benzene in [3+2] Cycloaddition Reactions. J Mol Graph Model 102:107760. https://doi.org/10.1016/j.jmgm.2020.107760
Mohammad-Salim Haydar A, Ahmed Basheer H, Abdallah HH, Zeroual A, Abdi Jamila L (2021) A molecular electron density theory study for [3+2] cycloaddition reactions of N-benzylcyclohexylnitrone with methyl-3-butenoate. New J Chem 45:262–267. https://doi.org/10.1039/D0NJ04049E
Salah M, Zeroual A, Jorio S, El Hadki H, Kabbaj O, Marakchi K, Komiha N (2020) Theoretical study of the 1,3-DC reaction between fluorinated alkynes and azides: Reactivity indices, Transition structures, IGM and ELF analysis. J Mol Graph Model 94:107458. https://doi.org/10.1016/j.jmgm.2019.107458
Salah M, Belghiti M. E, Aitouna A.O, Zeroual A, Jorio S, El Alaoui Abdellaoui H, El Hadki H, Marakchi K, Komiha N (2021) MEDT Study of the 1,3-DC Reaction of Diazomethane with Psilostachyin and investigation about the interactions of some pyrazoline derivatives with Protease (Mpro) of nCoV-2. J Mol Graph Model 102:107763. https://doi.org/10.1016/j.jmgm.2020.107763
Aitouna AO, Barhoumi A, Zeroual A (2023) A Mechanism Study and an Investigation of the Reason for the Stereoselectivity in the [4+2] Cycloaddition Reaction between Cyclopentadiene and Gem-substituted Ethylene Electrophiles. Sci Radices 2(3):217–228 https://doi.org/10.58332/scirad2023v2i3a01
Zeroual A, Ríos-Gutiérrez M, El idrissi M, El Alaoui El Abdallaoui H, Domingo Luis R (2019) An MEDT study of the mechanism and selectivities of the [3+2] cycloaddition reaction of tomentosin with benzonitrile oxide. Int J Quantum Chem 1–9. https://doi.org/10.1002/qua.25980
Ouled Aitouna A, Barhoumi A, El Alaoui El Abdallaoui H, Mazoir N, Elalaoui Belghiti M, Syed A, Bahkali AH, Verma M, Zeroual A (2023) Explaining the selectivities and the mechanism of [3+2] cycloloaddition reaction between isoalantolactone and diazocyclopropane. J Mol Model. https://doi.org/10.1007/s00894-023-05688-0
El Ghozlani M, Barhoumi A, Elkacmi R, Ouled Aitouna A, Zeroual A, El Idrissi M (2020) Mechanistic Study of Hetero-Diels–Alder [4 + 2] Cycloaddition Reactions Between 2-Nitro-1H-Pyrrole and Isoprene. Chemistry Africa 3:901–909. https://doi.org/10.1007/s42250-020-00187-8
Zeroual A, Ríos-Gutiérrez M, Amiri O, El idrissi M, Domingo LR (2019) An MEDT study of the mechanism, chemo and stereoselectivity of the epoxidation reaction of R-carvone with peracetic acid, RSC Advances. R Soc Chem 9:28500–28509. https://doi.org/10.1039/c9ra05309c
Aitouna AO, Belghiti ME, Eşme A, Anouar E, Aitouna AO, Zeroual A, Salah M, Chekroun A, El Abdallaoui HEA, Benharref A, Mazoir N (2021) Chemical reactivities and molecular docking studies of parthenolide with the main protease of HEP-G2 and SARS-CoV-2. J Mol Struct 130705. https://doi.org/10.1016/j.molstruc.2021.130705
Ouled Aitouna Ab, Barhoumi A, El idrissi M, Ouled Aitouna A, Zeroual A, Mazoir N, Chakroun A, Benharref A (2021) Theoretical investigation of the mechanism, chemo- and stereospecifity in the epoxidation reaction of limonene with meta-chloroperoxybenzoic acid (m-CPBA). Mor J Chem 9(1):75–82. https://doi.org/10.48317/IMIST.PRSM/morjchem-v9i1.20462
Ouled Aitouna Ab, Belghiti M.E, Eşme A, Ouled Aitouna An, Salah M, Chekroun A, El Alaoui El Abdallaoui H, Benharref A, Mazoir N, Zeroual A, Nejjari C (2021) Divulging the regioselectivity of epoxides in the ring-opening reaction, and potential himachalene derivatives predicted to target the antibacterial activities and SARS-CoV-2 spike protein with docking study. J Mol Struct 1244:130864. https://doi.org/10.5267/j.ccl.2023.3.008
Zoubir M, Belghiti ME, El Idrissi M (2022) Theoretical investigation of the mechanism and regioselectivity of the 3-isopropyl-1,6-dimethyl-naphthalene and ar-himachalene in nitration reaction: a MEDT study. Theor Chem Acc 141:8. https://doi.org/10.1007/s00214-022-02869-7
Zahnoune R, Asserne F, Ourhriss N, Aitouna AO, Barhoumi A, Hakmaoui Y, Belghiti ME, Abouricha S, Zeroual A (2022) Theoretical survey of Diels-Alder between acrylic acid and isoprene catalyzed by the titanium tetrachloride and titanium tertafluoride. J Mol Struct 1269:133630. https://doi.org/10.1016/j.molstruc.2022.133630
Rouani M, Rahmouni B, Chammache M, El-Mebtoul A, Ilidrissi A (2014) Comparative study of reactivity of (-)-R-carvone, (-)-R-linalool and (-)-(1S,4S)-camphor derivatives: Synthesis of new heterocycles. Eur J Chem 5(3):383–387. https://doi.org/10.5155/eurjchem.5.3.383-387.1029
McLean AD, Chandler GS (1980) Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z=11-18. J Chem Phys 72:5639–5648. https://doi.org/10.1063/1.438980
Raghavachari K, Binkley JS, Seeger R, Pople J (1980) A Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions. J Chem Phys 72:650–654. https://doi.org/10.1063/1.438955
Gaussian 09, Revision A.02, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li M, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian, Inc., Wallingford CT
Schlegel HB (1982) Optimization of equilibrium geometries and transition structures. J Comput Chem 2:214e218. https://doi.org/10.1002/wcms.34
Schlegel HB, Yarkony DR (1994) Modern Electronic Structure Theory, World Scientific Publishing, Singapore. https://doi.org/10.48419/IMIST.PRSM/rhazes-v9.21253
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of MethodsBased on continuous distributions of the solvent. Chem Rev 94:2027–2094
Cossi M, Barone V, Cammi R, Tomasi J (1996) Ab initio study of solvated molecules: anew implementation of the polarizable continuum model. Chem Phys Lett 255:327–327. https://doi.org/10.1016/0009-2614(96)00349-1
Cances E, Mennucci B, Tomasi J (1997) Evaluation of solvent effects in isotropic and Anisotropic dielectrics and in ionic solutions with a unified integral equation method:theoretical bases, computational implementation, and numerical applications. J Chem Phys 101:10506e10517. https://doi.org/10.1021/JP971959K
Barone V, Cossi M, Tomasi J (1998) Geometry optimization of molecular structures insolution by the polarizable continuum model. J Comput Chem 19:404e417
Parr RG, Szentpaly LV, Liu S (1999) Electrophilicity Index. J Am Chem Soc 121:1922e1924. https://doi.org/10.1021/ja983494x
Domingo L.R, Chamorro E, Prerez P (2008) Understanding the reactivity of CaptodativeEthylenes in polar cycloaddition reactions. A theoretical study. J Org Chem 73:4615e4624. https://doi.org/10.1021/jo800572a
Domingo LR, Perez P, Séaez JA (2013) Understanding the local reactivity in polar organicreactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486e1494. https://doi.org/10.1039/C2RA22886F
Zeroual A, Zoubir M, Benharref A, El Hajbi (2015) A Understanding the regioselectivity and reactivity of Fridley Crafts benzoylation using Parr function. Moroc J Chem 3:356–360. https://doi.org/10.48317/IMIST.PRSM/morjchem-v3i4.3050
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic andmolecular systems. J Chem Phys 92:5397e5403. https://doi.org/10.1063/1.458517
Noury S, Krokidis X, Fuster F, Silvi B (1999) Computational tools for the electron localizationfunction topological analysis. Comput Chem 23:597–604. https://doi.org/10.1016/S0097-8485(99)00039-X
Karmil F, Bounouar N, Mountadar S, Rich A, Belghiti ME, Zeroual A, Mountadar M (2022) Surf Interfaces 35:102438. https://doi.org/10.1016/j.surfin.2022.102438
Biovia DS (2017) "Materials studio" R2, Dassault Systèmes BIOVIA, San Diego
Song Z, Yanfeng XuL, Bao ZL, Pin Y, Yajin Q, Zhu H, Zhao W, Han Y, Qin C (2019) From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses 11:59. https://doi.org/10.3390/v11010059
Mali SN, Sawant S, Chaudhari HK, Mandewale MC (2019) Current Computer - Aided Drug Design 15:445–455(11). https://doi.org/10.2174/1573409915666190206142756
Mali SN, Anand A, Zaki MEA, Al-Hussain SA, Jawarkar RD, Pandey A, Kuznetsov A (2023) Theoretical and Anti-Klebsiella pneumoniae Evaluations of Substituted 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamide and Imidazopyridine Hydrazide Derivatives. Molecules 28(6):2801. https://doi.org/10.3390/molecules28062801
Raji H, Aitouna AO, Barhoumi A, Chekroun A, Zeroual A, Syed A, Elgorban AM, Verma M, Benharref A, Varma RS (2023) Antiviral docking analysis, semisynthesis and mechanistic studies on the origin of stereo- and chemoselectivity in epoxidation reaction of α′-trans-Himachalene. J Mol Liq 385:122204. https://doi.org/10.1016/j.molliq.2023.122204
Asserne F, Ouahdi Z, Hakmaoui Y et al (2023) Molecular Docking, Regio, Chemo and Stereoselectivity Study of the [3 + 2] Cycloaddition Reaction Between Pyridazi-3-one and Nitrilimine. Chemistry Africa. https://doi.org/10.1007/s42250-023-00735-y
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ (1998) Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J Comput Chem 19:1639–1662. https://doi.org/10.1002/(SICI)1096-987X(19981115)19:14%3c1639::AID-JCC10%3e3.0.CO;2-B
Schrödinger L, DeLano W (2020) PyMOL. Retrieved from http://www.pymol.org/pymol
Karmil FZ, Bounouar N, Mountadar S, Rich A, Belghiti ME, Zeroual A, Mountadar M (2022) Phosphate removal from contaminated seawater and RO water using magnetically modified reactive CaO derived from reject brine: Experimental studies maintained by theoretical simulations. Surf Interfaces 35:102438. https://doi.org/10.1016/j.surfin.2022.102438
Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSP2023R367), King Saud University, Riyadh, Saudi Arabia.
Funding
The authors extend their appreciation to the Researchers Supporting Project number (RSP2023R367), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Ali Barhoumi, Kamal Riyachi, Mohammed Elalaoui Belghiti and Abdessamad Tounsi: Article writing. Asad Syed and Mohammed Chafi: Conceptualization, Methodology; Ling Shing Wong revising and editing language; Discussion; Mohammed El idrissi, and Abdellah Zeroual Final review and editing. (All authors: analysis and interpretation of data and drafting the article).
Corresponding author
Ethics declarations
Ethics Approval
The manuscript is prepared in compliance with the Ethics in Publishing Policy as described in the Guide for Authors.
Conflicts of Interest
The authors declare that they have no conflict of interest and no competing interests exist.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Barhoumi, A., Ryachi, K., Belghiti, M.E. et al. Chromatography Scrutiny, Molecular Docking, Clarifying the Selectivities and the Mechanism of [3 + 2] Cycloloaddition Reaction between Linallol and Chlorobenzene-Nitrile-oxide. J Fluoresc 34, 1913–1929 (2024). https://doi.org/10.1007/s10895-023-03411-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-023-03411-z