
Abstract
Recently, bimetallic clusters have attracted a great deal of attention from research community because clusters yield intriguing properties ranging from the molecular and the bulk materials, which have extensive applications in nanomaterials. Clusters with tailored properties are governed by cluster size, geometrical structures, and elemental composition. Motivated by that we systematically investigated the structural, relative stable, and electronic properties of PbnSbn (n = 2–12) clusters by means of density functional theory. In this paper, the ground state structures, average binding energies, fragmentation energies, HOMO–LUMO gaps, and density of states were theoretically calculated. The results demonstrate that the large clusters adopt distorted ellipsoid structures with no symmetry. The average binding energies tend to be stable when cluster size n ≥ 4. Pb5Sb5 and Pb9Sb9 clusters are more chemically stable compared with the neighboring PbnSbn clusters, which may serve as the cluster assembled materials. The density of states of PbnSbn (n = 2–12) clusters moving toward more negative energy levels with the growing cluster size n, which also becoming more nonlocalized as the clusters size n increasing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The main investigations of cluster science are to investigate the essential properties of clusters and promising applications of clusters in nanomaterials, which making an essential bridge between the molecules and macro materials [1,2,3]. During the past several decades, the pure clusters composed of group IV or group V elements have been highly investigated by scholars from all over the world. As an example, the growth behaviors and fundamental properties of Cn clusters [4,5,6], Sin clusters [7, 8], Gen clusters [9, 10], Snn clusters [11, 12], Pbn clusters [13, 14], Asn clusters [15, 16], and Sbn clusters [17, 18] have been systematically investigated by both experiments and DFT calculations. Although many investigations have proved that the pure clusters or binary clusters from the same group of the periodic table elements have many similar properties [19,20,21,22] since the similar electronic configurations of the elements and analogous geometric structures of clusters. However, the investigations of bimetallic clusters are of great interest and significance due to the valence electron configurations of the both elements are different, thus bimetallic clusters may yield intriguing properties which show huge differences to both of the pure clusters.
Pb is the group IV element while Sb is the group V element. Therefore, they are the particularly evident p-block elements. The semiconductor materials consist of group III–V elements have contributed to the modern electronics, since scientist had discovered C60 which have exploited a new material field in carbon nanotubes and fullerenes. The discovery of cluster assembled materials with tunable properties by fine-tuning of element composition and geometrical structures are significant. It is well known that clusters are different from their molecules and condensed matters. Moreover, the physical and chemical properties of clusters are different with their size n, geometrical structures, and chemical composition. Therefore, it is crucial to elucidate the relationship between the clusters’ properties and their cluster size n for a given chemical composition system.
So far, Pbn clusters, Pb-based clusters, Sbn clusters, and Sb-based clusters have been systematically investigated. For Pbn clusters, the clusters with small cluster size n adopt planar structures while large clusters possess three dimensional structures. In addition, Pbn clusters demonstrate a metal-to-nonmetal transition with growing cluster size n. The cluster formation and ionization of Pbn clusters were analyzed by Saito et al. [23] using the time-of-flight mass spectrometer. Rajesh et al. [24] investigated the geometrical structures and electronic structures of Pbn and Pb +n (n = 2–15) cluster using density functional theory. Sascha et al. [25] carried out a molecular beam experiment to study the electric deflection of Pbn (n = 7–38) clusters. Senz et al. [26] applied the core–hole photoelectron spectroscopy as a probe to study lead clusters, it was found that lead clusters demonstrate a metal-to-nonmetal transition which is in well accordance with density functional theory calculations. For Sbn clusters, in the vapor phases, Sb tends to form tetrahedral clusters which are similar to the P4 cluster, As4 cluster. Polak et al. [1] investigated Sb−, Sb2−, Sb3−, and Sb4− clusters by photoelectron spectra. Zhou et al. [20] theoretically calculated the structural and electronic properties of Sbn (n = 2–10) clusters within density functional theory. Bernhardt et al. [19] investigated the decomposition mechanisms of Sb +n (n = 3–12) clusters versus cluster size n, surface type, as well as collision energy using surface collision induced dissociation mass spectrometry. In terms of bimetallic Pb-based and bimetallic Sb-based clusters, Rajesh et al. [27] theoretically investigated PbnM (M = C, Al, In, Mg, Sr, Ba, and Pb; n = 8, 10, 12, and 14) clusters, in order to investigate the effects of impurity M atoms on structural and electronic properties Pbn clusters through comparing the differences between Pbn clusters and PbnM clusters. Chen et al. [28] investigated the structural and magnetic properties of MPb10 and [MPb10]2 (M = Fe, Co, and Ni) clusters using density functional theory. Steinert et al. [29] synthesized the Mn/Sb clusters in the silicon matrix in order to explore the formation of MnSb clusters.
There are some experimental investigations on the growth pattern and content intensity properties of PbSb clusters [21, 22, 30,31,32,33]. According to the experimental results involving mass-spectrometer-based methods [32, 33], the PbSb, Pb2Sb2, Pb3Sb3, and Pb4Sb4 clusters were observed, demonstrating that the bimetallic PbnSbm clusters with equal molar ratio of Pb atoms and Sb atoms were found with enhanced stability. Furthermore, the binding energies of clusters were detected by photoelectron spectroscopy [21]. However, the PbnSbn (n > 4) clusters were not achieved by experiments. It is believed that the larger clusters are difficult to be generated compared with the smaller clusters, even the larger cluster are difficult to be detected by experimental devices owing to the limited resolution of instruments [34].
Density functional theory has been proved to be an efficient and powerful tool to investigate the physical and chemical properties of clusters, because the calculated results can be in well agreement with the experimental ones [34, 35]. Although some bimetallic clusters composed of Pb and Sb have been investigated using experiments and DFT calculations, there are scarce data for the PbnSbn clusters larger than Pb4Sb4 cluster. In this paper, the structural and electronic properties of PbnSbn (n = 2–12) have been investigated by means of density functional theory. We firmly believe that the fundamental investigation of PbnSbn clusters is interesting and vital, which may exceed to find cluster-assembled nanomaterials and novel properties.
Computational Methods
In the present work, all calculations were performed using DFT method, as implemented in the DMol3 code [20, 36, 37]. The ground state structures of PbnSbn clusters were achieved by the following steps. Firstly, the starting structures of PbnSbn clusters were built based on the ground state structures of Pbn−1Sbn−1 clusters. The structures of PbnSbn clusters were extensively searched by ab initio molecular dynamics, where time step is 1 fs, total simulation time is 100 ps. Secondly, following geometrical optimizations were performed based on the local minimal energy structures of ab initio molecular dynamics. Thirdly, the energy calculations were carried out based on the optimized structures. Finally, the ground state structures of PbnSbn clusters can be obtained through comparing the energies of the different structures of PbnSbn clusters. The singlet spin was used for the PbnSbn clusters with even numbers of electrons, whereas the double spin was used for the PbnSbn clusters with odd numbers of electrons during geometrical optimizations. Because the higher spin state, the higher energy of the structure was obtained. The spin unrestricted and formal spin as initial was used during energy calculations and ab initio molecular dynamics. The PBE functional within the general gradient approximation (GGA) was employed to treat exchange and correlation interactions [20, 28, 38, 39]. The DFT-semi-core potential was used to replace the internal electrons of PbnSbn systems. The basis set is double numerical basis combined with polarized functions (DNP). The SCF calculations were performed till the change of total energy is less than 10−6 Ha. The convergence standards of force and energy: the maximum force is less than 10−5 Ha Å−1, the energy per atom is converged to 10−5 Ha Å−1, and the maximum displacement is converged to 0.005 Å. The charge and spin involving density mixing standard are 0.2 and 0.5, respectively. The smearing (0.005 Ha) was used in order to achieve good convergence results. In addition, the direct inversion in iterative subspace (DIIS) method was also used in order to speed up the calculations and reduce calculation costs [40, 41]. The frequency calculations were performed to ensure that the lowest energy structures of PbnSbn (n = 2–12) clusters are located on the true minima of the corresponding potential surfaces. The parameters of Sb2 and Pb2 dimers were calculated in order to confirm the reliability of GGA–PBE functional for PbnSbn systems. In this work, the calculated bond length, vibrational frequency, total binding energy, and vertical ionization potential of Sb2 dimer are 2.574 Å, 260.52 cm−1, 2.82 eV, and 8.18 eV, respectively, while the corresponding experimental data are 2.49 Å [42], 270 cm−1 [43], and 3.1 eV [19], and 9.275 eV [43]. The calculated bond length, vibrational frequency, and vertical ionization potential of Pb2 dimer are 2.961 Å, 119.65 cm−1, 6.46 eV, respectively, and the experimental data are 2.93 Å [36], 110 cm−1 [36], 6.2 eV [23], respectively. It is obvious that the calculated parameters excellently agree with the experimental ones. Therefore, the calculation methods in this work are reliable.
Results and Discussion
Ground State Structures and Vibrational Spectra of PbnSbn (n = 2–12) Clusters
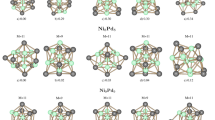
Here, the lowest energy structures of PbnSbn (n = 2–12) clusters were focused, because all the properties of PbnSbn clusters were calculated based on the lowest energy structures. All the structures are the energetic minima structures because the lowest energy structures were verified with no imaginary frequency. The lowest energy structures of PbnSbn (n = 2–12) clusters are displayed in Fig. 1, and the atomic coordinates of lowest energy structures of PbnSbn (n = 2–12) clusters are shown in Tables S1–S11 (the details can be seen in the supporting information). As it is shown in Fig. 1, the Pb2Sb2 cluster favors a triangular pyramid structure, and its point group symmetry is C2. It has been shown that Pb4 cluster adopts a planar structure [44], whereas Sb4 cluster also forms a triangular pyramid structure [20] which is similar to the geometrical structure of Pb2Sb2 cluster. Pb3Sb3 cluster forms a structure which a Pb atom and a Sb atom bind together and capped on the same side of the parallelogram. In addition, Pb3Sb3 cluster exhibits Cs symmetry. For Pb4Sb4 cluster, a distorted bilayer parallelogram with no symmetry is the most stable structure of Pb4Sb4 cluster. Pb5Sb5 cluster is composed of two parts, one part is a distorted pentagonal pyramid, and the other part is a distorted quadrangle. The point group symmetry of Pb5Sb5 cluster belongs to Cs. The ground state structure of Pb6Sb6 cluster is similar to that of Pb5Sb5 cluster, shows point group symmetry of C2. Pb7Sb7 cluster with Cs symmetry is a cage-like structure, and most of the surfaces show distorted quadrangle structures and rest of the surfaces show triangle structures. Pb8Sb8 cluster possesses C1 point group symmetry. The ground state structure of Pb8Sb8 cluster likes a distorted ellipsoid structure. It is interesting to point out that PbnSbn (n = 9–12) clusters also adopt ellipsoid structures, which show no symmetry. According to the Jahn–Teller distortion theorem, the lower symmetry can lower the energies of the clusters [45,46,47]. Therefore, it may be the reason why the large clusters with no symmetry are energetically favorable. On the basis of the growth pattern of PbnSbn (n = 2–12) clusters, the large clusters tend to adopt distorted ellipsoid structures and prefer no symmetry.

The lowest energy structures of PbnSbn (n = 2–12) clusters. The corresponding point group symmetry is shown in the parentheses. The purple ball is Sb atom, and the gray ball is Pb atom
Here, the vibrational spectra of PbnSbn (n = 2–12) clusters were calculated in order to provide the comparison for experimental results, because the cluster structures can be determined by comparing the calculated spectra and experimental spectra, and the calculated results would provide additional theoretical information for the experimental results. The vibrational spectra of PbnSbn (n = 2–12) clusters are shown in Fig. S1 (the details can be seen in the supporting information). The vibrational frequencies of PbnSbn clusters are in the range of 0–250 cm−1. The location of vibrational frequencies, the relative intensities of vibrational peaks, as well as the shapes of vibrational peaks are the important signals to identify the structures of PbnSbn clusters.
Relative Stable and Electronic Properties of PbnSbn (n = 2–12) Clusters
The stable properties of PbnSbn (n = 2–12) clusters were calculated on the basis of the lowest energy structures. The binding energy per atom (Eb) of PbnSbn cluster can be calculated using the formula [48,49,50,51]
where E(Pb), E(Sb), E(PbnSbn) are the energies of Pb atom, Sb atom, and PbnSbn cluster, respectively. Hence, the average binding energy is a good index to describe the thermodynamic properties of PbnSbn cluster. Figure 2 shows the average binding energies of PbnSbn clusters as a function of cluster size n. Firstly, average binding energies increase sharply with the increasing size n when cluster size n are in the range of n = 2–4, then average binding energies approach stable when size n ≥ 4. Therefore, the geometrical structures of PbnSbn (n = 2–12) clusters tend to be stable when cluster size n ≥ 4.

The average binding energies of PbnSbn (n = 2–12) clusters as a function of cluster size n
The fragmentation energy (\({\Delta}\text{E}\)) can demonstrates the relative stabilities of PbnSbn clusters compared with their neighboring clusters. The definition of \({\Delta}\text{E}\) can be defined as [52]
where E(Pbn−1Sbn−1), E(PbSb), and E(PbnSbn) represent the total energies of the Pbn−1Sbn−1 cluster, PbSb cluster, and PbnSbn cluster, respectively. \({\Delta}\text{E}\) as a function of cluster size n for PbnSbn clusters is shown in Fig. 3. The fragmentation energies show that there are some obvious oscillating behaviors. Several local peaks are found at n = 4, 6, 9, and 11, revealing that Pb4Sb4 cluster, Pb6Sb6 cluster, Pb9Sb9 cluster and Pb11Sb11 cluster are more thermodynamically stable compared with the neighboring PbnSbn clusters. In general, the fragmentation energies show a decrease trend as the cluster size n increasing.

The fragmentation energies of PbnSbn (n = 2–12) clusters as a function of cluster size n
In order to analyze the chemical stabilities of PbnSbn clusters, the HOMO–LUMO gaps of PbnSbn (n = 2–12) clusters were calculated. The HOMO–LUMO gap is a significant parameter to reflect the chemical stability of a cluster, which demonstrates the energy gap between the highest occupied orbital and the lowest unoccupied orbital [53]. A higher value of HOMO–LUMO gap represents that the cluster is more chemically stable, whereas a smaller value of HOMO–LUMO gap exhibits that the cluster is more chemically unstable. The HOMO–LUMO gaps of PbnSbn (n = 2–12) clusters as a function of cluster size n is shown in Fig. 4. From the Figure, we can see that there are obvious oscillations within the cluster size range of n = 2–12. In general, the trend of HOMO–LUMO gaps decrease with the increasing cluster size n. The two local maxima of HOMO–LUMO gaps are found at n = 5 and n = 9. The local maxima of HOMO–LUMO gaps of PbnSbn clusters suggest that Pb5Sb5 cluster and Pb9Sb9 cluster are chemically stable compared with the neighboring PbnSbn clusters. Furthermore, Pb5Sb5 cluster and Pb9Sb9 cluster have great potential in cluster assembled materials due to their chemical stabilities.

The HOMO–LUMO gaps of of PbnSbn (n = 2–12) clusters as a function of cluster size n
In order to well understand the electronic properties of PbnSbn (n = 2–12) clusters, the density of states of PbnSbn (n = 2–12) clusters were calculated, as shown in Fig. 5. In the present work, the density of states near fermi levels were focused because those density of states near the fermi levels are significant. From the Figures, the obvious trend of density of states of PbnSbn clusters can be observed, which density of states move toward more negative energy levels with increasing cluster size n. In other words, the fermi levels shift to more positive energy levels with increasing cluster size n. The most positive energy levels of conduction bands tend to be stable and approach stable at 0.08 Ha. For the density of states near the fermi levels, the contributions of p electrons are the maximum, the contributions of s electrons are the minimum, and that of d electrons are in between p electrons and s electrons. Hence, the p electrons have the largest contributions to the fermi levels, which may be originated from that the Pb and Sb elements are the evident p-block elements. For the density of states in the range of − 0.5 ~ − 0.2 Ha, the s and p electrons almost have the same contributions, whereas d electrons have the smallest contributions. For the density of states in the range of 0.01–0.4 Ha, the p and d electrons have the equal contributions while s electrons have smallest contributions. Moreover, density of states becoming more nonlocalized with the growing clusters size n, demonstrating that the metallicity of PbnSbn clusters becoming more predominant with the growing clusters size n.

Density of states of PbnSbn (n = 2–12) clusters. The fermi level is viewed as a vertical dash line, and set to zero
Conclusions
In the present work, the structural, relative stable and electronic properties of PbnSbn (n = 2–12) clusters have been systematically investigated in the frame work of density functional theory. The PbnSbn clusters with large size n prefer distorted ellipsoid structures and demonstrate no symmetry. The average binding energies of PbnSbn clusters tend to be stable when cluster size n ≥ 4. On the basis of fragmentation energies, Pb4Sb4 cluster, Pb6Sb6 cluster, Pb9Sb9 cluster and Pb11Sb11 cluster are more thermodynamically stable compared with their neighboring PbnSbn clusters. Pb5Sb5 cluster and Pb9Sb9 cluster are more chemically stable based on the HOMO–LUMO gap analysis. The density of states of PbnSbn (n = 2–12) clusters shift toward more negative energy levels with the growing cluster size n, and density of states becoming more nonlocalized with the increasing cluster size n.
References
M. L. Polak, G. Gerber, J. Ho, and W. C. Lineberger (1992). J. Chem. Phys. 97, 8990–9000.
H. Yang, Y. Wang, X. Chen, X. Zhao, L. Gu, H. Huang, J. Yan, C. Xu, G. Li, J. Wu, A. J. Edwards, B. Dittrich, Z. Tang, D. Wang, L. Lehtovaara, H. Häkkinen, and N. Zheng (2016). Nat. Commun. 7, 12809.
I. M. L. Billas, A. Châtelain, and W. A. de Heer (1994). Science 265, 1682–1684.
O. A. Van and R. J. Saykally (1998). Chem. Rev. 98, 2313–2357.
L. Belau, S. E. Wheeler, B. W. Ticknor, M. Ahmed, S. R. Leone, W. D. Allen, and M. A. Duncan (2007). J. Am. Chem. Soc. 129, 10229–10243.
J. Hutter, H. P. Luethi, and F. Diederich (1994). J. Am. Chem. Soc. 116, 750–756.
M. Haertelt, J. T. Lyon, P. Claes, H. J. De, P. Lievens, and A. Fielicke (2012). J. Chem. Phys. 136, 114.
K. D. Rinnen and M. L. Mandich (1992). Phys. Rev. Lett. 69, 1823–1826.
S. Heiles, S. Schäfer, and R. Schäfer (2011). J. Chem. Phys. 135, 034303.
W. Qin, W. C. Lu, Q. J. Zang, L. Z. Zhao, G. J. Chen, C. Z. Wang, and K. M. Ho (2010). J. Chem. Phys. 132, 214509.
S. Schäfer, B. Assadollahzadeh, M. Mehring, P. Schwerdtfeger, and R. Schäfer (2008). J. Phys. Chem. A 112, 12312.
B. Assadollahzadeh, S. Schäfer, and P. Schwerdtfeger (2010). J. Comput. Chem. 31, 929–937.
H. Li, Y. Ji, F. Wang, S. F. Li, Q. Sun, and Y. Jia (2011). Phys. Rev. B 83, 075429.
X.-P. Li, W.-C. Lu, Q.-J. Zang, G.-J. Chen, C. Z. Wang, and K. M. Ho (2009). J. Phys. Chem. A 113, 6217–6221.
R. K. Yoo, B. Ruscic, and J. Berkowitz (1992). J. Chem. Phys. 96, 6696–6709.
X. Bai, Q. Zhang, A. Gao, and J. Yang (1009). Comput. Theor. Chem. 2013, 94–102.
X. Zhou, J. Zhao, X. Chen, and W. Lu (2005). Phys. Rev. A 72, 053203.
R. O. Jones, O. Ahlstedt, J. Akola, and M. Ropo (2017). J. Chem. Phys. 146, 1291–12100.
T. M. Bernhardt, B. Kaiser, and K. Rademann (2002). Phys. Chem. Chem. Phys. 4, 1192–1200.
X. Zhou, J. Zhao, X. Chen, and W. Lu (2005). Phys. Rev. A 72, 053203.
J. J. Melko, U. Werner, R. Mitric, V. Bonacic-Koutecky, and A. W. Castleman Jr. (2011). J. Phys. Chem. A. 115, 10276–10280.
D. Schild, R. Pflaum, G. Riefer, and E. Recknagel (1988). Zeitschrift Für Physik D Atoms Molecules & Clusters 10, 329–335.
S. Yahachi, Y. Kenzi, M. Kazuhiro, and N. Tamotsu (1982). Jpn. J. Appl. Phys. 21, L396.
C. Rajesh and C. Majumder (2007). J. Chem. Phys. 126, 244704.
S. Schafer, S. Heiles, J. A. Becker, and R. Schafer (2008). J. Chem. Phys. 129, 044304.
V. Senz, T. Fischer, P. Oelssner, J. Tiggesbaumker, J. Stanzel, C. Bostedt, H. Thomas, M. Schoffler, L. Foucar, M. Martins, J. Neville, M. Neeb, T. Moller, W. Wurth, E. Ruhl, R. Dorner, H. Schmidt-Bocking, W. Eberhardt, G. Gantefor, R. Treusch, P. Radcliffe, and K. H. Meiwes-Broer (2009). Phys. Rev. Lett. 102, 138303.
C. Rajesh and C. Majumder (2008). J. Chem. Phys. 128, 024308.
X. Chen, K. Deng, C. Xiao, J. Chen, and D. E. Ellis (2011). Comput. Theor. Chem. 971, 73–76.
M. Steinert, W. Wesch, A. Undisz, M. Rettenmayr, W. Nunes, R. Borges, M. Godinho, R. Rubinger, M. Carmo, and N. Sobolev (2008). J. Phys. D: Appl. Phys. 42, 035406.
R. W. Farley, P. Ziemann, and A. W. C. Jr (1989). Zeitschrift Für Physik D Atoms Molecules & Clusters 14, 353–360.
R. Wheeler, K. LaiHing, W. Wilson, J. Allen, R. King, and M. Duncan (1986). J. Am. Chem. Soc. 108, 8101–8102.
K. F. Willey, K. Laihing, T. G. Taylor, and M. A. Duncan (1993). J. Phys. Chem. 97, (29), 7435–7440.
D. Schild, R. Pflaum, K. Sattler, and E. Recknagel (1987). J. Phys. Chem. 91, 2649–2653.
J. J. Melko, S. V. Ong, U. Gupta, J. U. Reveles, J. D’Emidio, S. N. Khanna, and A. W. Castleman (2010). Phys. Chem. C 114, 20907–20916.
E. C. Honea, A. Ogura, C. A. Murray, K. Raghavachari, W. O. Sprenger, M. F. Jarrold, and W. L. Brown (1993). Nature 366, 42–44.
B. Wang, J. Zhao, X. Chen, D. Shi, and G. Wang (2005). Phys. Rev. A 71, 309–315.
M. Bo, Y. Wang, Y. Huang, W. Zhou, C. Li, and C. Q. Sun (2014). J. Mater. Chem. C 2, 6090.
B. Song, W. Jiang, B. Yang, X. Chen, B. Xu, L. Kong, D. Liu, and Y. Dai (2016). Metall. Mater. Trans. A 47, 5214–5222.
J. Deng, Y. Lei, S. Wen, and Z. Chen (2015). Int. J. Miner. Process. 140, 43–49.
G. L. Zhang, H. K. Yuan, H. Chen, A. L. Kuang, Y. Li, J. Z. Wang, and J. Chen (2014). J. Chem. Phys. 141, 244304.
M. Zhang, L.-M. He, L.-X. Zhao, X.-J. Feng, and Y.-H. Luo (2009). J. Phys. Chem. C 113, 6491–6496.
G. Gerber and G. Kuscher (1981). Chem. Phys. 60, 119–131.
R. K. Yoo, B. Ruscic, and J. Berkowitz (1993). J. Chem. Phys. 99, 8445–8450.
C. Rajesh, C. Majumder, M. G. R. Rajan, and S. K. Kulshreshtha (2005). Phys. Rev. B 72, 235411.
M. E. Eberhart, R. C. O’Handley, and K. H. Johnson (1984). Phys. Rev. B 29, 1097–1100.
X. Li, B. Kiran, L.-F. Cui, and L.-S. Wang (2005). Phys Rev. Lett. 95, 253401.
Y.-R. Zhao, X.-Y. Kuang, B.-B. Zheng, Y.-F. Li, and S.-J. Wang (2011). J. Phys. Chem. A 115, 569–576.
K. O. Alcantar-Medina, M. Herrera-Trejo, A. Tlahuice-Flores, S. Martinez-Vargas, J. Oliva, and A. I. Martinez (1099). Comput. Theor. Chem. 2017, 55–63.
D. Toprek and V. Koteski (1081). Comput. Theor. Chem. 2016, 9–17.
Y. Jin, G. Maroulis, X. Kuang, L. Ding, C. Lu, J. Wang, J. Lv, C. Zhang, and M. Ju (2015). Phys. Chem. Chem. Phys. 17, 13590.
X. X. Xia, A. Hermann, X. Y. Kuang, Y. Y. Jin, C. Lu, and X. D. Xing (2016). J. Phys. Chem. C 120, 677–684.
S. Safer, S. Mahtout, K. Rezouali, M. A. Belkhir, and F. Rabilloud (1090). Comput. Theor. Chem. 2016, 23–33.
W. G. Sun, J. J. Wang, C. Lu, X. X. Xia, X. Y. Kuang, and A. Hermann (2017). Inorg. Chem. 56, 1241–1248.
Acknowledgements
This work was supported by the Regional Foundation of the NSFC (51664032), General Program of the NSFC (51474116), Program of China Scholarships Council (No. 201808530022), Joint Foundation of the NSFC-Yunnan province (U1502271), Cultivating Plan Program for the Leader in Science and Technology of Yunnan Province (2014HA003), Program for Nonferrous Metals Vacuum Metallurgy Innovation Team of Ministry of Science and Technology (2014RA4018), National Key Research and Development Program of China (2016YFC0400404), Youth Program of NSFC (51504115) and Program for Innovative Research Team in University of Ministry of Education of China (IRT_17R48), Science and Technology Talent Cultivation Plan of Yunnan Province, China (2017HB009).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, G., Chen, X., Yang, H. et al. The Density Functional Theory Investigation on the Structural, Relative Stable and Electronic Properties of Bimetallic PbnSbn (n = 2–12) Clusters. J Clust Sci 29, 1305–1311 (2018). https://doi.org/10.1007/s10876-018-1450-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-018-1450-y