
Abstract
The structural, relative stable and electronic properties of PbnSnn (n = 2–12) alloy clusters were systematically studied using density functional theory. The isomers of PbnSnn alloy clusters were generated and determined by ab initio molecular dynamics. By comparing the calculated parameters of Pb2 dimer and Sn2 dimers with the parameters from experiments, our calculations are reasonable. With the lowest-energy structures for PbnSnn clusters, the average binding energies, fragmentation energies, second- order energy differences, vertical ionization potentials, vertical electron affinities, HOMO–LUMO gaps, and density of states were calculated and analyzed. The results indicate that the Sn atoms have a tendency to bond together, the average binding energies tend to be stable up to n = 8, Pb8Sn8 cluster is a good candidate to calculate the molecular interaction energy parameter in Wilson equation, the clusters become less chemical stable and show an insulator-to-metallic transition, 3, 6, 8 and 11 are magic numbers of PbnSnn (n = 2–12) clusters, the charges always transfer from Sn atoms to Pb atoms in PbnSnn clusters except for Pb10Sn10 cluster, and density of states of PbnSnn clusters becoming continuous and shifting toward negative with the increasing size n.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Clusters demonstrate excellent catalytic [1, 2], adsorbent [3, 4], magnetic [5, 6], sensing [7, 8] and optical [9, 10] properties, etc. The investigation of clusters is a fundamental study but it’s very important and interesting, because the fundamental investigation can accelerate our steps on understanding the novel properties of cluster-assembled materials at atomic scale [11, 12]. As we all know, the properties of nanomaterials depend on its micro-structures and electronic structures essentially [13]. Then, the investigations of structural and electronic properties of clusters on particular materials are essential, which may facilitate the design of new materials with novel properties. However, some difficulties ahead of us, determining the structural and electronic properties of nanomaterials by experiments are of great difficult [14]. But the study on theoretical calculation is a good method due to the calculated results can provide reasonable results compared with the experimental data at a certain extent. Currently, the structural, relative stable and electronic studies of alloy clusters using density functional theory have attracted many scholar’s attention from all over the world.
Pb is harmful to human health and legislations have been established by government to prevent people from Pb. However, the Pb-containing solders are widely used in the world, which poses great threat to people’s health. The low-cost and accessible Pb-free solders are receiving increased attention from the material community. Then, the properties of PbnSnn clusters would provide very important information for the Pb-containing solders and the materials to replace harmful Pb in Pb-containing solders. Moreover, the candidate PbnSnn clusters can be used to calculate the molecular interaction energy parameters in the Wilson equation which can be used to calculate the phase diagram of vapor–liquid equilibria (VLE) [15, 16].
The pure Snn clusters and Pbn clusters were investigated extensively. With the experimental and theoretical investigations on Snn Clusters and Pbn clusters, the results show that these clusters show insulator-to-metallic transitions [17,18,19,20]. As the size n of Snn clusters and Pbn clusters increasing, the properties of those clusters convergence to the bulk state [21]. Osmekhin et al. [17] applied the photoelectron spectroscopy to study free nanoscale Snn clusters in the size n ranged from a few tens to 500 atoms, and the metal-to-semiconductor transition was discovered. Majumder et al. [22,23,24] systematically investigated ground-state structures of neutral and cation Snn clusters (n ≤ 20), the results show that growth behavior of Snn clusters is different from Sin clusters and Gen clusters at n ≥ 8 but some of them are similar, and the vertical ionization potentials and fragmentation behavior of Snn clusters are in accordance with experimental results. Bachels et al. [25] determined a formation enthalpy per atom of Snn clusters with the average size of n = 150. Saito et al. [26] investigated the relative abundance of Pbn clusters using time-of-flight mass spectrometer and electron bombardment, the results show the size n of the clusters up to ten atoms were observed and Pb7 cluster shows the highest intensity. Tchaplyguine et al. [18] and Bahn et al. [19] applied 4f and 5d photoelectron spectroscopy to investigate neutral and anionic Pbn clusters are in the range of 10–100, the results show that with the increasing size n of Pbn clusters, the non-metallic to metallic transition was observed. Whereas, Wang et al. [20] systematically calculated the Pbn (n = 2–22) clusters using DFT, the covalent to metallic transition was also found. Majesh et al. [27] investigated the atomic and electronic properties of Pbn (n = 2–15) and Pb +n (n = 2–15) clusters by density functional theory, the results show that the cationic and neutral clusters show similar growth patterns, and 4, 7, 10 and 13 are the magic numbers. Li et al. [28] calculated the neutral Pbn (n = 21–30) clusters using first principle calculation, the results show that Pb22–26 clusters favor endohedral cage structures and Pb27–30 clusters prefer three endohedral structures, and 24 and 28 are the magic numbers.
The Sn-based alloy clusters and Pb-based alloy clusters were also aroused much interest due to the properties of the alloy clusters are different from the pure metallic clusters. Sosa-Hernández et al. [29] reported the lowest-energy isomers and magnetic properties and stabilities of CoxSny clusters (x + y ≤ 5) using density functional theory, they found that the geometries are significantly different to the pure clusters, the magnetic behavior is independent of the concentration of Co and the mixing of bimetallic clusters are favorable. Samanta et al. [30] studied the structures, binding and electronic properties of SnmGen (m + n ≤ 5) clusters with density functional theory, the results would provide some fundamental theory for the mechanism of CO oxidation on SnmGen clusters. Sosa-Hernández et al. [31] adopted density functional theory to study the structural and electronic properties of FexSny (x + y ≤ 5) clusters, they found that the binary clusters are different from the pure clusters, the magnetic properties are not related with the Fe concentration. Tai et al. [32] reported small SnnZn (n = 1–12) with DFT and CCSD methods, the results show that impurity Zn atom on the surface of Snn clusters, the Zn atom is encapsulated into the empty cages of Snn clusters with n = 11 and n = 12, the transition appears at n = 9 and n = 10, and Sn12Zn cluster and Sn10Zn cluster are the magic clusters. Paz-Borbón et al. [33] investigated hydrogenation of C2H4 to C2H6 over RuN clusters and (RuSn)N clusters with density functional theory, the results show that the (RuSn)N clusters have pronounced effects on the hydrogenation compared with RuN clusters, RuN clusters adopt cubic conformations while (RuSn)N clusters prefer compact Ru cores capped by Sn atoms. Melko et al. [34] studied the structural and electronic properties of PbxSb −y clusters and SnxBi −y clusters combined photoelectron spectroscopy with DFT, the results show that the clusters may not be the excellent building blocks,but qualified for the applications in energetic, electronic and optical function. Xing et al. [35] investigated the magic numbers of (M/Pb)− (M = Au, Ag and Cu) clusters by laser ablation and ToF-MS, they found that most of the magic clusters can be explained by electron shell models. Melko et al. [36] investigated the structural and electronic properties of neutral and anionic PbxIny (x ≤ 5, y ≤ 7) clusters by photoelectron spectroscopy and DFT, the results show that stabilities of some clusters were enhanced, which can provide valuable guidances for cluster-assembled materials. Rajesh et al. [37] systematically investigated the effects of impurity atoms on PbnM (M = C, Al, In, Mg, Sr and Ba) clusters with first principle calculations, the results reveal that the location of impurity atom on PbnM cluster depends on atomic radius of the impurity atom and the interactions between the host cluster and the impurity atom, Mg atom lowers the stabilities of Pbn clusters while C and Al enhance the stability. Barman et al. [38] systematically studied the structural and electronic properties of Pbn−1Sn clusters and Snn−1Pb clusters with first principle calculations, the results show that dopant Pb atom lowers the stabilities of Snn−1Pb clusters while dopant Sn atom enhances the stabilities of Pbn−1Sn clusters, 4, 7, 10 and 12 are the magic numbers of Snn−1Pb clusters and 4, 7 and 9 are the magic numbers of Pbn−1Sn clusters. Orel et al [39] studied the geometries, relative energies and electronic properties of global minima of SnmPbn (7 ≤ m + n ≤ 12) clusters, generally, the calculated results are excellent agreement with experimental data.
Although the structural and electronic properties of Snn clusters, Pbn clusters, Sn-based clusters and Pb-based clusters were extensive investigated by density functional theory and experimental investigations. The PbnSnn clusters were scarcely reported, we believe that our work is meaningful. In this paper, we will focus our attention on the structural, relative stable and electronic properties of PbnSnn (n = 2–12) clusters using density functional theory.
Computational Methods
In this work, all the density functional theory calculations were carried out in DMol3 package [20, 40]. In order to ignore the effects of the initio structures, the isomers of PbnSnn clusters were extensively generated by ab initio molecular dynamics. During the dynamics calculation, the temperature maintains at 300 K, 1 ps for each time step, total simulation time is 100 ps. The GGA [40]-BLYP [20] was used to treat the exchange-correlation functional. The core was treated by DFT semi-core potential. Basis set was treated by double numerical basis with polarized functions (DNP) [20, 40]. The different spin multiplicities were taken into consideration in the geometric optimization processes. A SCF convergence criterion is 10−6 Ha on the total energy. With respect to density mixing standard, 0.2 and 0.5 are for charge and spin, respectively. In order to obtain a good convergence result, the smearing was used and its value is 0.005 Ha. Energy convergence is 10−5 Ha per atom and that of maximum force is 0.002 Ha/Å. The maximum displacement is less than 0.005 Å.
The parameters of Pb2 dimer and Sn2 dimer were calculated to verify the computational schematic processes, whereas the experimental parameters of the dimers were achieved from literature, the detailed information are shown in Table 1. Comparing the calculated and experimental data, we can clearly see that the calculated data are excellent agreement with experimental data. It is obvious that our computational methods are reliable.
Results and Discussion
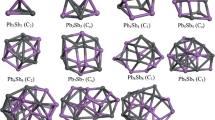
The ground-state structures of PbnSnn clusters are shown in Fig. 1. From Fig. 1, we can see that the Pb2Sn2 cluster is in the shape of distorted parallelogram which two Sn atoms on the same side, and its point group symmetry is Cs. The ground-state structures of Pb4 cluster and Sn4 cluster are also parallelogram structures [21, 43]. The Pb3Sn3 cluster is a bipyramid with C2v symmetry, the three Sn atoms on the same plane. It is interesting to note that the Pb2Sn2 cluster is a planar structure while the Pb3Sn3 cluster is a three dimensional structure, the PbnSnn clusters show a planar structure to three dimensional structure transition. Additionally, the similar transition was also observed in Snn clusters from Sn4 cluster to Sn6 cluster and that of Pbn clusters from Pb4 cluster to Pb6 cluster [21, 43]. The Pb4Sn4 cluster is an inclined four prism without any symmetry. Furthermore, the PbnSnn clusters are don’t show any symmetry from n = 4 to n = 12, which could reduce the total energies of the clusters and make the clusters more stable [44]. The structure of Pb5Sn5 cluster is similar with that of Pb4Sn4 cluster by capped one Pb atom and one Sn atom. Pb6Sn6 cluster can be regarded as a bi-layer structure. From n = 7 to n = 12, the shapes of the clusters are ellipsoid and the positions of Pb atoms and Sn atoms are out of order, but the Sn atoms tend to gather together. In addition, the characteristic frequencies show a oscillator for PbnSnn clusters in the range of n = 2–8, whereas for n = 8–12, the characteristic frequencies decrease with increasing size n except for Pb12Sn12 Cluster. This may be related to the ground state structures of PbnSnn (n = 2–12) clusters.

Ground-state structures and point groups of PbnSnn (n = 2–12) clusters (corresponding characteristic frequencies are in the parentheses)
The binding energies per atom of lowest-energy structures of PbnSnn clusters can be defined as [39, 45,46,47,48,49,50,51]
where E(Sn), E(Pb) and E(PbnSnn) are energies of Sn atom, Pb atom and PbnSnn clusters, respectively. The average binding energies (Eb) of PbnSnn (n = 2–12) clusters are shown in Fig. 2. Binding energy can reflect the thermodynamic stabilities of clusters. From Fig. 2, the average binding energies increase with the increasing size n, then it tend to be stable up to n = 8. The similar trend of binding energies were also observed in pure Snn clusters [24] and pure Pbn clusters [20]. It suggests that the thermodynamic stabilities of PbnSnn clusters increase and the stabilities tend to be stable when n up to 8. It is concluded that PbnSnn (n ≥ 8) clusters are the ideal candidates to calculate the molecular interaction energy parameter in phase diagram of vapor–liquid equilibria (VLE) [15]. The slope between Pb2Sn2 cluster and Pb3Sn3 cluster shows steep increase which means the compactness of Pb3Sn3 cluster are better than Pb2Sn2 cluster. In addition, it is originated from the transition from a planar structure to a three dimensional structure.

Average binding energies of PbnSnn (n = 2–12) clusters
The fragmentation energies (ΔE) and second-order energy differences (Δ2E) can be defined as [46, 52,53,54]
where E(PbSn)n−1, E(PbSn), E(PbSn)n and E(PbSn)n+1 represent the energies of Pb n−1Snn−1 cluster, PbSn cluster, PbnSnn cluster and Pbn+1Snn+1 cluster, respectively. The ΔE and Δ2E of PbnSnn clusters are presented in Figs. 3 and 4. From Fig. 3, the ΔE of Pb3Sn3 cluster, Pb8Sn8 cluster and Pb11Sn11 cluster have larger values than their neighboring clusters, indicating that the three clusters are more stable than their neighbors. From Fig. 4, the trend of second-order energy differences is excellent agreement with that of second-order energy differences. We also find that Pb3Sn3 cluster, Pb8Sn8 cluster and Pb11Sn11 cluster are higher stabilities than their neighbors.

The ΔE of PbnSnn (n = 2–12) clusters

The Δ2E of PbnSnn (n = 2–12) clusters
The vertical ionization potentials of PbnSnn clusters are shown in Fig. 5. Vertical ionization potential is defined as the neutral cluster loses one electron and adsorbs energy to become a positive charged cluster, and the energy can be viewed as the vertical ionization potential [51, 52]. It means that the higher value of vertical ionization potential, the more difficult for the neutral cluster to ionize and becomes a positive cluster. As we can see from Fig. 5, the values of vertical ionization potentials decreasing with the variation of size n, which also show that the PbnSnn clusters become more and more easier to lose one electron and become a positive cluster. Based on the trend, we can refer that metallicity of PbnSnn clusters become more and more stronger.

Vertical ionization potentials (VIP) of PbnSnn (n = 2–12) clusters
Vertical electron affinities of PbnSnn clusters are shown in Fig. 6. The vertical electron affinity can reflect the chemical stabilities of the neutral clusters [51, 52]. The higher value of vertical electron affinity, the easier for a neutral cluster to accept one electron and becomes a negative cluster. As it is shown in Fig. 6, the vertical electron affinities of PbnSnn clusters increase with increasing size n, which means the neutral clusters become more and more easier to accept electrons and become negative clusters.

Vertical electron affinities (VEA) of PbnSnn (n = 2–12) clusters
The HOMO–LUMO gaps of PbnSnn clusters are shown in Fig. 7. The HOMO–LUMO gap (HL gap) can also reflect the chemical stabilities of the clusters [53, 54]. The higher HL gap means the gap between highest occupied molecular orbitals and lowest unoccupied molecular orbitals are larger, which indicates that it is difficult for electrons transfer from conduction band to valence band, and the neutral cluster is more chemically stable. From Fig. 7, there is a general trend that values of HL gaps sharply decrease with the variation of size n, which means the chemical stabilities of PbnSnn clusters also sharply decrease and the trend is excellent agreement with that of VIPs and VEAs. It is also referred that the PbnSnn clusters show a covalent-to-metallic transition in the range of n = 2–12. Moreover, Pure Pbn clusters and pure Snn clusters also show covalent-to-metallic transitions [17, 18]. Compared the transition size n of pure Pbn clusters and pure Snn clusters, the alloy PbnSnn clusters show the transition with smaller size n. It is interesting to note that the values of Pb3Sn3 cluster, Pb6Sn6 cluster, Pb8Sn8 cluster and Pb11Sn11 cluster are larger than their neighbors, which means that the Pb3Sn3 cluster, Pb6Sn6 cluster, Pb8Sn8 cluster and Pb11Sn11 cluster are more chemically stable than their neighbors and are also excellent agreement with that of the clusters analyzed by VIPs and VEAs. Furthermore, we conclude that 3, 6, 8 and 11 are magic numbers of PbnSnn (n = 2–12) clusters.

HOMO–LUMO gaps of PbnSnn (n = 2–12) clusters
The average Mülliken charges are shown in Fig. 8. According to the results, the charges always transfer from Sn atoms to Pb atoms except for Pb10Sn10 cluster, which Pb atoms act as electron acceptors while Sn atoms act as electron donators. This situation is similar with that of Sn atoms to Pt atoms in PtnSnn clusters [40] and Al atoms to Ni atoms in NinAln clusters [46]. In addition, it is originated from the negativities of element Sn and element Pb (1.96 for Sn and 2.33 for Pb in terms of Pauling’s electronegativity scale). There is a trend that the charges decrease with increasing size n, and Pb10Sn10 cluster at the turning point, the charges show an odd-even transition up to n = 6 that the clusters with the odd number atoms have less charges transfer than that of the clusters with the even number atoms. It is referred that the PbnSnn clusters may be good catalysts. With respect to the binary Pb–Sn nano-catalysts, the Pb atoms have redundant electrons and would act as active site, whereas those of Sn atoms protect the Pb atoms [40]. From the Fig. 8, Pb3Sn3 cluster, Pb5Sn5 cluster and Pb8Sn8 cluster are located at the local peaks, which imply the strong interactions between Pb atoms and Sn atoms in Pb3Sn3 cluster, Pb5Sn5 cluster and Pb8Sn8 cluster, and they are agreement with the results of fragmentation energies, second-order energy differences and HL gaps.

Average Mülliken charges analysis of PbnSnn (n = 2–12) clusters
In order to further explore the electronic properties of PbnSnn clusters, we plotted the density of states of PbnSnn clusters, the details are shown in Fig. 9. The Fermi level is plotted as a vertical dash line and shift to zero. From Fig. 9, we can see that the density of states are mainly consist of two parts, the density of states are between −0.4 to −0.14 Ha and −0.1 to −0.3 Ha, respectively. The density of states between −0.4 and 0.14 Ha are contributed by s electrons and p electrons, which s electrons and p electrons almost have the same contributions, while the density of states between −0.1 and 0.3 Ha are contributed by s electrons and p electrons and the contributions within the intervals are mainly contributed by p electrons. Moreover, it is interesting to note that the continuities of the density of states becoming better with the increasing size n, and they shift toward negative and tend to be stable, it may be the reason why the transition from insulator-to-metallic and the clusters become less chemical stable.

Density of states of PbnSnn (n = 2–12) clusters
Conclusions
We have systematically studied the structural, relative stable and electronic properties of PbnSnn clusters (n = 2–12) using density of states. All the calculations were carried out in DMol3 package at GGA-BLYP level. According the ground-state structures of PbnSnn clusters, the Sn atoms tend to bond together, and the transition from 2D structure to 3D structure was found. The average binding energies increase with increasing size n, and tend to be stable up to n = 8. Pb8Sn8 cluster is a good candidate to calculate the molecular interaction energy parameter for PbSn alloy system. The results of fragmentation energies, second-order energy differences and HOMO–LUMO gaps show that the Pb3Sn3 cluster, Pb6Sn6 cluster, Pb8Sn8 cluster and Pb11Sn11 cluster are more stable than their neighbors. The vertical ionization potentials increase with increasing size n while the vertical electron affinities show an opposite trend. The PbnSnn clusters show a covalent-to-metallic transition, and 3, 6, 8 and 11 are the magic numbers of PbnSnn clusters. The charges always transfer from the Sn atoms to Pb atoms except for Pb10Sn10 cluster based on the Mülliken charge analysis, which indicate the PbnSnn clusters may be ideal nano-catalysts with controllable properties. The density of states of PbnSnn clusters becoming continuous and shifting toward negative with the increasing size n, which may be the reason why the clusters become less chemical stable and the transition from insulator-to-metallic.
References
C. T. Campbell (2013). The Energetics of Supported Metal Nanoparticles: Relationships to Sintering Rates and Catalytic Activity. Acc. Chem. Res. 46, 1712.
H. Zhang, M. Jin, Y. Xiong, B. Lim, and Y. Xia (2013). Shape-Controlled Synthesis of Pd Nanocrystals and Their Catalytic Applications. Acc. Chem. Res. 46, 1783.
D. Cortes-Arriagada, M. P. Oyarzun, L. Sanhueza, and A. Toro-Labbe (2015). Binding of Trivalent Arsenic onto the Tetrahedral Au20 and Au19Pt Clusters: Implications in Adsorption and Sensing. J. Phys. Chem. A 119, 6909.
S. Hirabayashi and M. Ichihashi (2015). NO Decomposition Activated by Preadsorption of O2 onto Copper Cluster Anions. J. Phys. Chem. C 119, 10850.
Z. Ben-Xia, D. Dong, W. Ling, and Y. Ji-Xian (2014). Density functional study on the structural, electronic, and magnetic properties of 3d transition-metal-doped Au5 clusters. J. Phys. Chem. A 118, 4005.
F. Aguilera-Granja, M. B. Torres, A. Vega, and L. C. Balbas (2012). Structural, electronic, and magnetic properties Of ConCum nanoalloys (m + n = 12) from first principles calculations. J. Phys. Chem. A 116, 9353.
P. Chen, Y. Li, J. Ma, J. Huang, C. Chen, and H. Chang (2016). Size-tunable copper nanocluster aggregates and their application in hydrogen sulfide sensing on paper-based devices. Sci. Rep. 6, 24882.
J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao, and R. P. Van Duyne (2008). Biosensing with plasmonic nanosensors. Nat. Mater. 7, 442.
W. Ma and F. Chen (2012). Optical and electronic properties of Cu doped Ag clusters. J. Alloys Compd. 541, 79.
S. J. Oldenburg, J. B. Jackson, S. L. Westcott, and N. J. Halas (1999). Infrared extinction properties of gold nanoshells. Appl. Phys. Lett. 75, 2897.
M. Zhou, C. Zeng, Y. Chen, S. Zhao, M. Y. Sfeir, M. Zhu, and R. Jin (2016). Evolution from the plasmon to exciton state in ligand-protected atomically precise gold nanoparticles. Nat. Commun. 7, 13240.
R. Jin, C. Zeng, M. Zhou, and Y. Chen (2016). Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities. Chem. Rev. 116, 10346.
O. Fenwick, E. Coutino-Gonzalez, D. Grandjean, W. Baekelant, F. Richard, S. Bonacchi, D. De Vos, P. Lievens, M. Roeffaers, J. Hofkens, and P. Samori (2016). Tuning the energetics and tailoring the optical properties of silver clusters confined in zeolites. Nat. Mater. 15, 1017.
G. Seifert (2004). Nanomaterials: Nanocluster magic. Nat. Mater. 3, 77.
X. Yang, X. Chen, C. Zhang, X. Xie, B. Yang, B. Xu, D. Liu, and H. Yang (2016). Prediction of vapor–liquid equilibria for the Pb–X (X = Ag, Cu and Sn) systems in vacuum distillation using ab initio methods and Wilson equation. Fluid Phase Equilib. 417, 25.
A. K. Sum and S. I. Sandler (1999). Use of ab initio methods to make phase equilibria predictions using activity coefficient models. Fluid Phase Equilib. 158–160, 375.
S. Osmekhin, M. Tchaplyguine, M. H. Mikkelä, M. Huttula, T. Andersson, O. Björneholm, and S. Aksela (2010). Size-dependent transformation of energy structure in free tin clusters studied by photoelectron spectroscopy. Phys. Rev. A 81, 023203.
M. Tchaplyguine, G. Öhrwall, T. Andersson, S. Svensson, O. Björneholm, M. Huttula, M. Mikkelä, S. Urpelainen, S. Osmekhin, A. Caló, S. Aksela, and H. Aksela (2014). Size-dependent evolution of electronic structure in neutral Pb clusters—As seen by synchrotron-based X-ray photoelectron spectroscopy. J. Electron Spectrosc. 195, 55.
J. Bahn, P. Oelßner, M. Köther, C. Braun, V. Senz, S. Palutke, M. Martins, E. Rühl, G. Ganteför, T. Möller, B. von Issendorff, D. Bauer, J. Tiggesbäumker, and K. H. Meiwes-Broer (2012). Pb 4f photoelectron spectroscopy on mass-selected anionic lead clusters at FLASH. New J. Phys. 14, 075008.
B. Wang, J. Zhao, X. Chen, D. Shi, and G. Wang (2005). Atomic structures and covalent-to-metallic transition of lead clusters Pbn (n = 2–22). Phys. Rev. A 71, 033201.
B. Assadollahzadeh, S. Schäfer, and P. Schwerdtfeger (2010). Electronic properties for small tin clusters Snn (n ≤ 20) from density functional theory and the convergence toward the solid state. J. Comput. Chem. 31, 929.
C. Majumder, V. Kumar, H. Mizuseki, and Y. Kawazoe (2001). Small clusters of tin: Atomic structures, energetics, and fragmentation behavior. Phys. Rev. B 64, 233405.
C. Majumder, V. Kumar, H. Mizuseki, and Y. Kawazoe (2002). Ionization potentials of small tin clusters: first principles calculations. Chem. Phys. Lett. 356, 36.
C. Majumder, V. Kumar, H. Mizuseki, and Y. Kawazoe (2005). Atomic and electronic structures of neutral and cation Snn (n = 2–20) clusters: A comparative theoretical study with different exchange-correlation functionals. Phys. Rev. B 71, 035401.
T. Bachels and R. Schäfer (1999). Formation enthalpies of Sn clusters: a calorimetric investigation. Chem. Phy. Lett. 300, 177.
S. Yahachi, Y. Kenzi, M. Kazuhiro, and N. Tamotsu (1982). Formation and Ionization Potentials of Lead Clusters. Jpn. J. Appl. Phys. 21, L396.
C. Rajesh and C. Majumder (2007). Atomic and electronic structures of neutral and charged Pbn clusters (n = 2–15): theoretical investigation based on density functional theory. J. Chem. Phys. 126, 244704.
X. Li, W. Lu, C. Wang, and K. M. Ho (2010). Structures of Pbn (n = 21–30) clusters from first-principles calculations. J. Phys. Condens. Matter 22, 465501.
E. M. Sosa-Hernández, J. M. Montejano-Carrizales, and P. G. Alvarado-Leyva (2015). Stability and magnetic behavior of small CoxSny (x + y ≤ 5) atomic clusters. J. Alloys Compd. 632, 772.
P. N. Samanta and K. K. Das (2012). Electronic structure, bonding, and properties of SnmGen (m + n ⩽ 5) clusters: A DFT study. Comput. Theor. Chem. 980, 123.
E. M. Sosa-Hernández, J. M. Montejano-Carrizales, and P. G. Alvarado Leyva (2015). Geometrical shapes, stabilities and electronic behavior of small FexSny (x + y ≤ 5) atomic clusters. Eur. Phys. J. D 69, 212.
T. B. Tai, N. M. Tam, and M. T. Nguyen (2011). Evolution of structures and stabilities of zinc-doped tin clusters SnnZn, n = 1–12. Three-dimensional aromaticity of the magic clusters Sn10Zn and Sn12Zn. Chem. Phys. 388, 1.
L. O. Paz-Borbon, A. Hellman, J. M. Thomas, and H. Gronbeck (2013). Efficient hydrogenation over single-site bimetallic RuSn clusters. Phys. Chem. Chem. Phys. 15, 9694.
J. J. Melko, U. Werner, R. Mitric, V. Bonacic-Koutecky, and A. W. Castleman Jr. (2011). Electronic structure similarities in PbxSb −y and SnxBi −y clusters. J. Phys. Chem. A 115, 10276.
X. Xing, Z. Tian, H. Liu, and Z. Tang (2003). Magic bimetallic cluster anions of M/Pb (M = Au, Ag and Cu) observed and analyzed by laser ablation and time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 17, 1411.
J. J. Melko, S. V. Ong, U. Gupta, J. U. Reveles, J. D’Emidio, S. N. Khanna, and A. W. Castleman (2010). Anion Photoelectron Spectroscopy and First-Principles Study of PbxIny Clusters. J. Phys. Chem. C 114, 20907.
C. Rajesh and C. Majumder (2008). Structure and electronic properties of PbnM (M = C, Al, In, Mg, Sr, Ba, and Pb; n = 8, 10, 12, and 14) clusters: theoretical investigations based on first principles calculations. J. Chem. Phys. 128, 024308.
S. Barman, C. Rajesh, G. P. Das, and C. Majumder (2009). Structural and electronic properties of Snn−1Pb and Pbn−1Sn clusters: a theoretical investigation through first principles calculations. Eur. Phys. J. D 55, 613.
S. Orel and R. Fournier (2013). Density functional theory and global optimization study of SnmPbn clusters (7 ⩽ m + n ⩽ 12, 0 ⩽ m/(m + n) ⩽ 1). J. Chem. Phys. 138, 064306.
X. Huang, Y. Su, L. Sai, J. Zhao, and V. Kumar (2015). Low-Energy Structures of Binary Pt–Sn Clusters from Global Search Using Genetic Algorithm and Density Functional Theory. J. Clust. Sci. 26, 389.
V. E. Bondybey, M. Heaven, and T. A. Miller (1983). Laser vaporization of tin: Spectra and ground state molecular parameters of Sn2. J. Chem. Phys. 78, 3593.
S. Yoshida and K. Fuke (1999). Photoionization studies of germanium and tin clusters in the energy region of 5.0–8.8 eV: Ionization potentials for Gen (n = 2–57) and Snn (n = 2–41). J. Chem. Phys. 111, 3880.
C. Rajesh, C. Majumder, M. G. R. Rajan, and S. K. Kulshreshtha (2005). Isomers of small Pbn clusters (n = 2–15): Geometric and electronic structures based onab initiomolecular dynamics simulations. Phys. Rev. B 72, 235411.
M. E. Eberhart, R. C. O’Handley, and K. H. Johnson (1984). Molecular-orbital models of structural phase transformations in crystalline and amorphous cobalt alloys. Phys. Rev. B 29, 1097.
Y. Bai, H. Cheng, H. Sun, N. Xu, and K. Deng (2011). Structures, stabilities and electronic properties of FePbn (n = 1–14) clusters: Density-functional theory investigations. Physica B 406, 3781.
J. Wen, J. Zhang, G. Chen, X. Zhang, and Z. Wen (2016). Structure, stability and magnetic properties of (NiAl)n(n ≤ 6) clusters. J. Phys. Chem. Solids 96–97, 68.
F. Suo, Y. Zhang, and S. Huang (2017). Theoretical Investigation of Electronic Properties of Undoped and Ag-Doped (CdTe)16×N Multi-cage Nanochains. J. Clust. Sci. 28, 1393.
K. Li, C. Yang, M. Wang, and X. Ma (2017). Adsorption and Dissociation of H2 on Cluster Al6N. J. Clust. Sci. 28, 1335.
Y. Jin, Y. Tian, X. Kuang, C. Zhang, C. Lu, J. Wang, J. Lv, L. Ding, and M. Ju (2015). Ab Initio Search for Global Minimum Structures of Pure and Boron Doped Silver Clusters. J. Phys. Chem. A 119, 6738.
X. X. Xia, A. Hermann, X. Y. Kuang, Y. Y. Jin, C. Lu, and X. D. Xing (2016). Study of the Structural and Electronic Properties of Neutral and Charged Niobium-Doped Silicon Clusters: Niobium Encapsulated in Silicon Cages. J. Phys. Chem. C 120, 677.
G. Li, J. Wang, X. Chen, Z. Zhou, H. Yang, B. Yang, B. Xu, and D. Liu (2017). Bimetallic PbnCun (n = 2–14) clusters were investigated by density functional theory. Comput. Theor. Chem. 1106, 21.
Y. Jin, G. Maroulis, X. Kuang, L. Ding, C. Lu, J. Wang, J. Lv, C. Zhang, and M. Ju (2015). Geometries, stabilities and fragmental channels of neutral and charged sulfur clusters: S Qn (n = 3–20, Q = 0, ±1). Phys. Chem. Chem. Phys. 17, 13590.
X. Xing, A. Hermann, X. Kuang, M. Ju, C. Lu, Y. Jin, X. Xia, and G. Maroulis (2016). Insights into the geometries, electronic and magnetic properties of neutral and charged palladium clusters. Sci. Rep. 6, 19656.
W. G. Sun, J. J. Wang, C. Lu, X. X. Xia, X. Y. Kuang, and A. Hermann (2017). Evolution of the Structural and Electronic Properties of Medium-Sized Sodium Clusters: A Honeycomb-Like Na20 Cluster. Inorg. Chem. 56, 1241–1248.
Acknowledgements
This work was supported by the Regional Foundation of the National Natural Science Foundation of China (51664032), the Foundation of the State Key Laboratory of Complex Nonferrous Metal Resources Clear Utilization (CNMRCUTS1503), the Joint Foundation of the National Natural Science Foundation of China–Yunnan province (U1502271), the Cultivating Plan Program for the Leader in Science and Technology of Yunnan Province (2014HA003), the Program for Nonferrous Metals Vacuum Metallurgy Innovation Team of Ministry of Science and Technology (2014RA4018) and the National Key Research and Development Program of China (2016YFC0400404).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, Gf., Zhou, Zq., Chen, Xm. et al. Structural, Relative Stable, and Electronic Properties of PbnSnn (n = 2–12) Clusters were Investigated Using Density Functional Theory. J Clust Sci 28, 2503–2516 (2017). https://doi.org/10.1007/s10876-017-1242-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-017-1242-9