
Abstract
In this work, the results of DFT-based calculations on the structures, stabilities, vibrational, electronic, and hydrogen storage behavior of (CaO) n rings are presented and discussed systematically. The equilibrium ring structures of Ca–O clusters for n = 2–5 are found to be stable. Vibrational frequencies and IR intensities further support the enhanced stability with an increase in the size of Ca–O clusters. The HOMO–LUMO surfaces and their derived parameters are used to explain the electronic properties of the titled systems. For efficient hydrogen storage, metals especially, the transition metals with large cohesive energy (CE) suffer from the problem of cohesion as it is expected that the adsorption energies of metal decorated absorbents should be larger than the CEs of metal. In order to avoid this, hydrogen adsorbed directly on the absorbents is preferred. Due to relatively smaller CEs of the s-block metals, hydrogen adsorbs directly on the cluster which indeed solves the problem of cohesion. The hydrogen storage capacity of (CaO) n clusters, considering hydrogen adsorption on (CaO)4 and (CaO)5 rings is studied. The outcomes appear to give meaningful and satisfactory results. Thus the present work is expected to lead further the applications of small clusters for easy, efficient, and eco-friendly hydrogen storage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is well known that calcium is placed directly below magnesium in the group IIA metals in the periodic table. Since Mg having 3 s valence electrons, the chemical properties of Ca can be investigated especially by the nature of its 4 s valence electrons. In spite of the fact that Ca–O and Mg–O clusters are supposed to show similar properties, appealing differences can also possible as calcium has a larger size and polarizability and a lower ionization potential and both the bulk metals as well as their oxides are more tightly bound that their Mg analogues. Apart from this, the experimental observations appear to indicate that small Ca–O clusters present stability trend different from those of clusters of the Mg–O frameworks.
There are several applications on small clusters having peculiar features which have been used enormously for intensive understanding of the building blocks of materials. Some properties of clusters are too unique to be found in their bulk analogues. For instance, metal oxide particles are often appraised to be bulk fragments but their features could be entirely different in small cluster systems [1,2,3]. Calcium oxide crystallizes in the rock-salt structure with mainly ionic bonding including some degree of covalency. This prototype oxide possesses a wide band-gap (~ 7 eV) and high dielectric constant (~ 12) [4]. Local density approximation based band-structure calculations provide a half-metallic ferromagnetic ground state for CaO [5]. CaO is used as catalyst, toxic-waste remediation agent, an additive in refractory, in the paint as well as for other fundamental applications [6]. CaO nanoparticles have also been promising as a destructive adsorbent for toxic chemical agent [7]. Nanocrystalline CaO is also widely used as an absorbent to remove COD from paper mill effluent.
Several experimental studies have been focused on the small clusters of Ca–O. The mass spectra of [(CaO) + n ] [8] and [(CaO) n Ca2+] [9] cluster ions have been reported and the experimental studies on singly and doubly charged clusters of Ca–O based on laser ionization time-of-flight mass spectrometry have also been reported [10]. Attempts have also been made to explain the general trends observed in these experiments. However, theoretical studies on Ca–O clusters are relatively scarce. Only a few futile attempts have been made to draw conclusion on the structures and bonding in Ca–O clusters [11, 12]. The present study is expected to enhance the understanding of structure and stability trends in Ca–O clusters. We have performed a systematic study on structures, stability [in terms of binding energy (BE) and Gibbs free energy of reaction], vibrational and electronic properties of (CaO) n ring clusters for n ≤ 5 employing a computational quantum mechanical modelling method as density functional theory (DFT) approach.
Hydrogen energy has been proven as a highly efficient and eco-friendly choice to fossil fuels [13, 14]. In practice, hydrogen requires being stored at optimum temperatures and pressures with fast kinetics and easy reversibility [15]. A natural alternative to adsorbent material would be materials having light elements. Hydrogen adsorption via carbon-based materials and their analogues have been extensively studied [16,17,18,19,20,21,22,23,24,25], however, some have been found to adsorb hydrogen very weakly [20,21,22]. The present work demonstrates hydrogen storage capability of (CaO) n ring clusters. For efficient hydrogen storage, the adsorption energies of metal decorated absorbents should be larger than the cohesive energies (CEs) of metal. Due to their large CEs, transition metals suffer from the problem of cohesion. To overcome this problem, hydrogen adsorbed directly on the absorbents should be preferred [26]. Since, s-block metals possess relatively smaller CEs [27], therefore, hydrogen adsorbs directly on the cluster which eliminates the problem of cohesion.
The goal of the present work is to provide a theoretical characterization of interactions in terms of BE (stability pattern) and the hydrogen storage behavior in the given clusters (CaO) n (n = 1–5). Aside from the theoretical study on the behavior of such kind of interactions and the hydrogen storage behavior, the structures, electronic features as well as the vibrational analysis have also been probed in the framework of the DFT approach which have also been highlighted in this work. To the best of our knowledge, this is the first report on the clusters derived from the Ca–O framework which deals with structural analysis including point group symmetry, electronic (HOMO–LUMO plots) and normal mode (vibrational assignments) features, analyzed from the DFT approach using B3LYP and M06-2X functionals with 6-311++G(d,p) basis set. The clusters of Ca–O framework obtained in the present study allow an extensive comparison with those obtained previously for Mg–O clusters [28]. Here, we report a systematic study of the results of an investigation of Ca–O clusters which is presented efficiently in the framework of DFT method with the additional support of the derived cyclic clusters (n = 4 and 5) representing the efficient hydrogen storage capacity behavior.
Computational Methods
The computations using DFT approach are based on generalized gradient approximated hybrid functional as the B3LYP which Becke’s three parameter with 20% Hartree–Fock (HF) exchange including the correlation term developed by Lee, Yang, and Parr [29, 30] and meta hybrid functional as M06-2X having functional (54% HF exchange) by Yan-Truhlar [31, 32] and the outcomes of the DFT approach are promising [33, 34] and they have been extensively used. A complete basis set, 6-311++G(d,p) which includes both diffuse as well as polarization functions has been exercised throughout the DFT calculations using B3LYP and M06-2X functionals which are a good choice for Ca–O clusters as reported by Bawa and Panas [35]. All geometries have been fully optimized with no symmetry constraint in the potential energy surfaces. Vibrational frequencies were also calculated at the same level of theory as aforementioned above. All quantum chemical computations were performed with the help of Gaussian 09 [36] package at Linux workstation. The calculated bond length (see Table 1) of CaO with C ∞v point group, 1.84 and 1.83 Å using the B3LYP and M06-2X functionals, respectively in which the bond length at M06-2X level of theory is in close proximity with the experimental outcome (1.82 Å) while the BE values for CaO monomer are 4.58 and 5.96 eV at B3LYP and M06-2X level of theory, correspondingly in which the estimated experimental BE value of the CaO monomer (4.76 eV) is close to the computed BE value (4.58 eV) using the B3LYP method [11, 12, 37]. The computed vibrational frequency (see Table 2) for CaO is 763.5 cm−1 which appear to give a general agreement with the experimental outcomes (732.1 cm−1) [37] as the DFT approach for CaO system has been in the gas phase. This may suggest the validity the DFT method employed here in this research work. The natural bond orbital (NBO) analyses have also been performed using the same aforementioned approach as mentioned above.
Results and Discussion
Structures and Stabilities
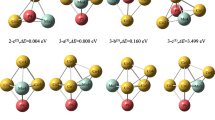
The equilibrium planar structures of (CaO) n small clusters (n = 2–5) can be discerned from Fig. 1. The cyclic ring clusters studied here (n = 1–5), strongly favor the higher order of symmetry with the increase in n. For example, here (CaO)4 tetramer planar cluster possesses D 4h point group of symmetry. More interestingly, they belong to at least some local minima in the potential energy surface. The structural parameters of planar (CaO) n clusters are listed in Table 1. Using the B3LYP method, one can note that the bond-length (Ca–O) increases from 1.84 Å (1.83 Å at M06-2X level of theory) in CaO (1.77 Å in case of Mg–O) to 2.06–2.07 Å in (CaO) n (n > 2) [1.83–1.89 Å in (MgO) n (n > 2)] and saturates which clearly shows that the Mg–O molecules are tightly bound to each other in Mg–O clusters. It can clearly be seen from Fig. 1 that in the case of ring clusters (n = 2–5) the ring size increases as the bond angles Ca–O–Ca and O–Ca–O increase. As the ring size increases, the geometrical parameters have been found to be in the same order using the both B3LYP and M06-2X approaches which can be reflected from the Table 1.

Optimized planar ring structures of (CaO) n rings at B3LYP and M06-2X levels
In order to analyze the energetics (stability pattern) of these planar (CaO) n clusters, we have also calculated other possible isomers of (CaO) n clusters as shown in Fig. 2. One can note that the planar (CaO) n ring structures correspond to global minima for n ≤ 3 but local minima for n ≥ 4. Few more insights regarding the feasibility of reaction (another parameter used for probing the stability order), the Gibbs free energy of formation (∆G form), n(CaO) → (CaO) n , has also been computed. The energy for the smallest ring cluster, (CaO)2 having C 2h point group, (see Fig. 1, system 2 in the Table 1), is depicted as − 4.66 and − 5.41 eV at B3LYP and M06-2X levels of study, respectively. For instance, a ladder isomer of n = 3 (see Fig. 2, ladder structure constituted by two squares, 3a in Table 2) has been found to be 0.10 eV and 0.03 eV higher in energy [i.e., relative energy (RE) with respect to the global minima structure] (less stable) than (CaO)3 ring cluster with C 2v point group at B3LYP and M06-2X levels of theory, respectively.

Isomers of (CaO) n clusters optimized at B3LYP and M06-2X levels of theory using 6-311++G(d,p) basis set with their energies relative to planar rings. Relative energies (B3LYP, M06-2X) are also given
In order to depict the stability of planar (CaO) n clusters, we have calculated the binding energy per CaO as follows;
where the total electronic energy of respective species including zero point correction is indicated by the term as E{..}. Tables 1 and 2 lists the calculated BE values of (CaO) n clusters, which suggest that the planar structures are stable against dissociation to CaO molecules.
The BE value for 3a ladder structure is 3.16 eV which fairly indicate that the ladder system is less stable than its ring cluster (3.19 eV) with the employment of the B3LYP method, which is strongly supported by the RE results. Surprisingly, the BE of (CaO)3 (see Fig. 1, species 3 from Table 1) (3.19 eV) is found to be more stable than the (MgO)3 cluster having BE 2.89 eV. The M06-2X method also gives the similar order of stability with the BE values of 3.64 eV for ladder isomer and 3.66 eV for (CaO)3 ring. The negative ∆G form values as − 8.62 and − 10.01 eV for n = 3 ring cluster clearly indicate that the reaction is more feasible (more stable) for the ring cluster than that of its isomer having values − 8.53 and − 9.95 eV which is in nice agreement with its BEs and RE values (see Table 1 for ring system and Table 2 for its isomer). As the polarity of a molecule depends on the electronegative difference, size, and charge of atoms, the order of polarity is CaO > MgO. The size of Mg2+ is smaller than Ca2+, due to this it has more covalent character than later that causes a decrease in polarity. On the contrary, it is interesting to note that in the case of n = 4 clusters, (CaO)4, the stability order using the BE, RE, and ∆G form parameters is: cube (BE: − 4.02 eV; RE: − 2.77 eV; ∆G form: − 14.38 eV) > ladder (BE: − 3.50 eV; RE: − 0.66 eV; ∆G form: − 12.51 eV) > ring (BE: − 3.33 eV; RE: 0.00; ∆G form: − 11.96 eV) at B3LYP level of study which resembles very well to relative each other as it is expected [see Fig. 1 for n = 4 ring possessing D 4h point group and Fig. 2 for its two isomers [cube like structure (species 4a in the Table 2) and ladder structure constituted by three squares (4b in the same Table)] which behavior is similar as in the case of Mg–O tetramer clusters. The M06-2X approach also gives the same stability pattern as cube (BE: − 4.69 eV; RE: − 3.51 eV; ∆G form: − 17.01 eV) > ladder (BE: − 4.04 eV; RE: − 0.92 eV; ∆G form: − 14.63 eV) > ring (BE: − 3.81 eV; RE: 0.00; ∆G form: − 13.81 eV) for n = 4 clusters as observed in the framework of the B3LYP method. In the case of (MgO)4, it should be noticed that (MgO)4 planar structure is 0.73 eV higher in energy than its global minima (cube-like structure).
Moreover, for n = 5, the optimized structures of the clusters of (CaO)5 have been displayed in the Fig. 1 (ring with D 5h point group) and Fig. 2 (three isomers of the ring cluster as 5a, 5b, and 5c given in the Table 2). Employing the B3LYP level of study, the stability pattern has been probed as: 5b (BE: − 3.98 eV; RE: − 3.00 eV; ∆G form: − 17.78 eV) > 5a (BE: − 3.69 eV; RE: − 1.55 eV; ∆G form: − 16.50 eV) > 5c (BE: − 3.63 eV; RE: − 1.22 eV; ∆G form: − 16.21 eV) which is also supported by the M06-2X level of theory with the appearance of the same stability order as: 5b (BE: − 4.61 eV; RE: − 3.82 eV; ∆G form: − 20.97 eV) > 5a (BE: − 4.27 eV; RE: − 2.05; ∆G form: − 19.30 eV) > 5c (BE: − 4.17 eV; RE: − 1.57 eV; ∆G form: − 18.90 eV).
Furthermore, the value of BE increases with the increase in n, which is in accordance to their decrement in the bond lengths. Therefore, their stability increases as there is an increment in the size (n) of (CaO) n rings. This is primarily because of the increase in the polarity of the Ca–O bond (vide infra).
Vibrational Properties
The calculation of vibrational properties provides further insight into the stability of equilibrium structures. The optimized structures with all real frequencies belong to true minima in the potential energy surfaces. It should be noted that our vibrational frequency calculations provide all real frequencies for (CaO) n clusters at both B3LYP and M06-2X levels. In order to focus on the vibrational characteristics of (CaO) n clusters, intense normal modes of vibrations have been listed in Table 3 along with their assignments.
It can be seen that vibrational frequencies and normal mode intensities increase with the enhancement in the size of the rings (n). For example, CaO stretching frequency increases from 621.5 cm−1 with intensity 338.5 a.u. in (CaO)2 to 698.9 cm−1 with intensity 1275.4 a.u. in (CaO)5 while in the case of Mg–O ring clusters, the vibrational frequency hikes from 654.2 cm−1 with intensity 148.4 a.u. and goes to 910.2 cm−1 with intensity 568.2 a.u. in (MgO)5 employing the same aforementioned computational method. The M06-2X functional also provide the normal mode of vibrations along with the intensity in the similar fashion as observed at B3LYP level of study. This observation is in accordance with the increase in the BE, hence the stability of (CaO) n clusters. The observations based on stretching frequency clearly shows that each Mg–O bond of all the rings clusters of MgO is stronger than their corresponding ring clusters of CaO. The lower stretching frequency of the (CaO)2 dimer system as compared to the CaO monomer system is consistent with its larger Ca–O bond distance. The frequencies corresponding to the in-plane bending of (CaO) n clusters decrease along with decrement in their corresponding intensities. On the contrary, it is nice to notice that out-of bending vibrations have nearly equal frequencies but their intensities increase with the increase in n. It is also to be noted that the frequencies of the twisting mode of (CaO) n clusters decrease slowly while their intensities enhance abruptly with the increase in the size of the cluster.
Electronic Properties
The highest occupied molecular orbitals (HOMOs) as well as lowest unoccupied molecular orbitals (LUMOs) of CaO monomer as well as (CaO) n (n = 2–5) planar ring clusters can be seen in Fig. 3. The HOMO, LUMO surfaces and their energy gap values have a pivotal role due to their orbitals (HOMO–LUMO) energy contribution and the contribution of orbitals on the molecular surface when they interact with the other molecule. It is interesting to notice that the contribution of p atomic orbitals of O is found in HOMOs and the LUMOs are contributed by s atomic orbitals of Ca which are in accordance with the results of the Mg–O clusters. The plotted molecular orbital energy spectra of (CaO) n (n = 2–5) planar ring systems can be seen from Fig. 4.

HOMO (upper set) and LUMO (lower set) surfaces of (CaO) n rings

Molecular orbital energy spectra of (CaO) n rings at B3LYP/6-311++G(d,p) level. Red and yellow lines correspond to occupied and unoccupied orbitals, respectively (Color figure online)
The chemical reactivity of (CaO) n can be analyzed in terms of HOMO–LUMO energy gap. The smaller HOMO–LUMO gap (HLG) corresponds to the more reactive (less stable) systems. The HLG of (CaO) n rings (listed in Table 4) suggests that (CaO)2 ring (the lowest HLG = 2.39/4.32 eV using B3LYP/M06-2X methods) should be most reactive whereas (CaO)3 ring with the highest HLG value 3.62 eV (least reactive) using the B3LYP method and (CaO)4 ring (the highest HLG value = 5.68 eV employing the M06-2X method) is the least reactive among all (CaO) n species. The similar outcomes have been observed in the case of Mg–O clusters (n = 2 and 3) at B3LYP level of study. The HLGs of CaO monomer and Ca–O clusters (n = 2–5) increases from n = 1 to n = 3(4) at B3LYP(M06-2X) level and then decreases at both levels of study which is illustrated in Table 4 while in the case of Mg–O clusters, the HOMO–LUMO energy gap values increase from n = 1–3 and then become almost constant from n = 3–5.
The negative of energies of HOMO and LUMO can be approximated by ionization potential (IP) and electron affinity (EA) within the framework of Koopmans’ theorem. The calculated IP and EA values of (CaO) n clusters are listed in Table 4. The IP values for the (CaO)3 ring (4.81 and 6.40 eV employing B3LYP and M06-2X methods, respecively) further suggests that it has least tendency to donate the electron i.e. less reactive. The EA of (CaO) n rings decreases with the increase in n from 2 to 5 using both levels of study. This also indicates the enhanced stability of (CaO) n clusters with the increase in n which is consistent with the increase in their BEs and the ∆G form values (see Table 1).
In order to analyze the charge distribution, we performed the NBO based population analysis. In a recent study, we have shown that NBO charges are more reliable when compared with various other population schemes [38]. The NBO charges on Ca and O atoms of (CaO) n clusters are also listed in Table 4. One can see that the NBO charges increase with an increase in the size of the ring, consequently increasing the polarity of the Ca–O bond. For instance, in (CaO)2 ring, there is a charge transfer of approximately + 1.52 e from each Ca to O atom while in the case of Mg–O dimer ring cluster, the NBO charge on the Mg has been reported as + 1.59 e which is greater than the NBO charge on Ca in (CaO)2 ring. The charge transfer increases from + 1.52 to + 1.67 e in (CaO) n (n = 2–5) cyclic clusters as can be discerned from Table 4. Using M06-2X functional, the NBO charges can also be verified from the Table 4 which is in accordance with the former B3LYP method. This increase in polarity of the Ca–O bond is responsible for the increase in the BE values of (CaO) n and hence their stability.
Hydrogen Storage Behavior
Calcium oxide (CaO) usually exhibits a higher activity among alkaline-earth metal oxides used in such processes because of a more delocalized electron distribution across the surface of oxygen atom of the CaO molecule [39]. Adsorption and reactivity study on CaO have been primarily employed on powders. As a result of the charge transfer in (CaO) n clusters (see Table 4), Ca–O bond begins to be polar which creates a local electric field and it is sufficient to polarize H2 molecules and hence bind them to the ring clusters. Since the cluster can adsorb the H2 molecule and release it based on the requirements. For optimum hydrogen storage, E ad (threshold value) equals 0.1–0.4 eV/H2 [15,16,17], implying that hydrogen can be adsorbed and desorbed easily under standard operating conditions.
We consider the case of (CaO)4 and (CaO)5 cyclic rings, as these rings are more polar and stable. The formula for adsorption energy (E ad) per H2 molecule is represented as;
where E{(CaO) n + m H2} represents the total electronic energy of (CaO) n (n = 4, 5) with adsorbed H2 molecules and m is the number of H2 molecules adsorbed.
The hydrogen adsorption mass ratio (w ad) has also been calculated as;
where M is the molecular weight of respective chemical species.
In order to get clear insights into the adsorption behavior of such ring clusters studied here, a number of H2 molecules are added one by one to the (CaO) n (n = 4 and 5) rings and the resulting structures have been reoptimized at the same level of theory. The procedure is rehearsed until the adsorption energy, E ad becomes comparable to 0.10 eV. After optimization of the successive interaction of Ca–O clusters with hydrogen molecules, we get a stable configuration of four hydrogen molecules adsorbed (CaO)4 and five H2 molecules adsorbed (CaO)5 cluster as displayed in Fig. 5. In the case of (CaO)4, the H2 molecules are polarized due to charge transfer effectively from O atom. One can note that the NBO charges on Ca and O atoms reduce to + 1.63e and − 1.60e from ± 1.65e in (CaO)4 ring. The distance of adsorbed H2 is 2.70 Å from Ca and 2.08 Å from O atom in (CaO)4 with the E ad of 0.12 eV per H2 however, in the case of (MgO)4 cluster, all H2 molecules have been adsorbed (E ad = 0.14 eV) by magnesium sites with the average distance 2.35 Å.

Equilibrium configuration of hydrogen adsorbed (CaO)4 and (CaO)5 rings. The distance of adsorbed molecules (black, in Å) and NBO charges (red, in e) are also displayed (Color figure online)
On the contrary, it is interesting to notice that in (CaO)5 ring cluster all hydrogen molecules are adsorbed by Ca sites with a distance of 2.72 Å which give the similar kind of H2 adsorption site as obtained in the case of (MgO)5 ring cluster with the average distance 2.36 Å. The atomic charges on each Ca atom reduce to + 1.64e from + 1.67e in free (CaO)5 ring cluster. The adsorption energy, E ad is found to 0.10 eV/H2 which is sufficiently large to confirm that hydrogen molecules remain physisorbed and they are feasible for the reversible adsorption phenomenon under optimum conditions. The percentage mass ratio of adsorbed hydrogen molecules by the Ca–O ring clusters has been calculated as 3.47% which is smaller than that of corresponding Be–O (7.5%) as well as Mg–O (4.76%) clusters which have been studied recently [28, 40]. The electronic properties and the hydrogen storage behavior (via the adsorption process) of the Mg–O clusters have been reported by the other research groups which also provide subtle as well as relevant information [41,42,43]. Very recently, a similar kind of computational study on planar Be–O pentamer ring cluster has been exhibited to adsorb five hydrogen molecules having adsorption energy 0.10 eV [40]. Thus, like Be–O and Mg–O clusters, (CaO) n clusters studied here, can also be used effectively for hydrogen storage which nowadays, is widely used.
Conclusions
In summary, a systematic study on structures, stabilities, vibrational, electronic, and hydrogen storage properties of (CaO) n clusters have been performed using B3LYP and M06-2X functionals having different percentage of HF exchange. The planar equilibrium structures for (CaO) n clusters (n = 2–5) were obtained and their stabilities were analyzed systematically. It has been noticed that the stability enhances with the increase in the size of clusters. The stability is found to be further enhanced with the increase in the contribution of HF exchange, i.e., from B3LYP to M06-2X functional. All fundamental modes of vibrations were assigned and observed trend of their vibrational frequencies and IR intensities have been discussed. Electronic properties of (CaO) n clusters are discussed by HOMO and LUMO surfaces as well as their other various derived electronic parameters such as ionization potential, electron affinity, etc. Finally, we have discussed the hydrogen adsorption on planar (CaO)4 and (CaO)5 ring clusters with adsorption energies 0.12 and 0.10 eV, respectively and compared our results with (MgO) n clusters which suggest easy, efficient, and eco-friendly hydrogen storage capability of (CaO) n and their future applications like Be–O and Mg–O clusters. From the above, it is concluded that the present study is yet another interesting recipe like Be–O as well as Mg–O clusters as to the utility, ability, and richness of the efficient, easy, and eco-friendly hydrogen storage behavior in general. Such works may assist researchers to design suitable nanoclusters having capability of effective hydrogen storage in the field of nanotechnology.
References
A. Jain, V. Kumar, M. Sluiter, and Y. Kawazoe (2006). Comput. Mater. Sci. 36, 171–175.
P. N. Kapoor, A. K. Bhagi, R. S. Mulukutla, and K. J. Klabunde Dekker Encyclopedia of Nanoscience and Technology (Marcel Dekker, New York, 2004).
A. Khaleel, P. N. Kapoor, and K. J. Klabunde (1999). NanoStruct. Mat. 11, 459–468.
R. C. Whited, C. J. Flaten, and W. C. Walker (1973). Solid State Commun. 13, 1903–1905.
I. S. Elfimov, S. Yunoki, and G. A. Sawatzky (2002). Phys. Rev. Lett. 89, 216403–216407.
B. K. Olga, L. Isabelle, and V. Alexander (1997). Chem. Mater. 9, 2468–2480.
O. Koper, X. L. Yong, and J. K. Kenneth (1993). Chem. Mater. 5, 500–505.
W. A. Saunders (1988). Phys. Rev. B 37, 6583–6586.
T. P. Martin and T. Bergmann (1989). J. Chem. Phys. 90, 6664–6667.
P. J. Ziemann and A. W. Castleman (1992). J. Phys. Chem. 96, 4271–4276.
F. Bawa and I. Panas (2002). Phys. Chem. Chem. Phys. 4, 103–108.
A. Vibok and G. J. Halasz (2001). Phys. Chem. Chem. Phys. 3, 3042–3047.
J. L. C. Rowsell and O. M. Yaghi (2006). J. Am. Chem. Soc. 128, 1304–1315.
A. M. Seayad and D. M. Antonelli (2004). Adv. Mater. 16, 765–777.
S. K. Bhatia and A. L. Myers (2006). Langmuir 22, 1688–1700.
Q. Sun, Q. Wang, P. Jena, and Y. Kawazoe (2005). J. Am. Chem. Soc. 127, 14582–14583.
Q. Sun, P. Jena, Q. Wang, and M. Marquez (2006). J. Am. Chem. Soc. 128, 9741–9745.
J. Zhou, Q. Wang, Q. Sun, and P. Jena (2011). J. Phys. Chem. C 115, 6136–6140.
T. Hussain, B. Pathak, T. A. Maark, C. M. Araujo, R. H. Scheicher, and R. Ahuja (2011). Europhys. Lett. 96, 27013–27016.
H. Dodziuk and G. Dolgonos (2002). Chem. Phys. Lett. 356, 79–83.
M. Shiraishi, T. Takenobu, and M. Ata (2003). Chem. Phys. Lett. 367, 633–636.
H. Kajiura, S. Tsutsui, K. Kadono, M. Kakuta, M. Ata, and Y. Murakami (2003). Appl. Phys. Lett. 82, 1105–1107.
C. Li, J. Li, F. Wu, S. S. Li, J. B. Xia, and L. W. Wang (2011). J. Phys. Chem. C 115, 23221–23225.
J. Li, Z. Hu, and G. Yang (2012). Chem. Phys. 392, 16–20.
H. Y. Wu, X. Fan, J. L. Kuo, and W. Q. Deng (2011). J. Phys. Chem. C 115, 9241–9249.
Y. Wang, X. Li, F. Wang, B. Xu, J. Zhang, Q. Sun, and Y. Jia (2013). Chem. Phys. 415, 26–30.
Y. S. Wang, Y. Ji, M. Li, P. F. Yuan, Q. Sun, Y. Jia (2011). J. Appl. Phys. 110, 094311/1-6.
A. K. Srivastava and N. Misra (2015). Mol. Simu. 42, 208–214.
A. D. Becke (1993). J. Chem. Phys. 98, 5648–5652.
A. Lee, W. Yang, and R. G. Parr (1988). Phys. Rev. B 37, 785–789.
Y. Zhao and D. G. Truhlar (2006). J. Chem. Phys. 125, 194101.
Y. Zhao and D. G. Truhlar (2008). Theor. Chem. Acc. 120, 215.
S. H. Mehdi, R. M. Ghalib, S. Awasthi, S. F. Alshahateet, R. Hashim, O. Sulaiman, and S. K. Pandey (2017). ChemistrySelect 2, 1–13.
S. K. Pandey, M. F. Khan, S. Awasthi, R. Sangwan, and S. Jain (2017). Aus. J. Chem. 70, 328–337.
F. Bawa and I. Pavas (2001). Phys. Chem. Chem. Phys. 3, 3042–3047.
J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox Gaussian 09, Rev B.01 (Gaussian Inc, Wallingford, 2010).
K. P. Huber and G. Herzberg Molecular Spectra and Molecular Structure, IV. Constants of Diatomic Molecules (van Nostrand Reinhold, New York, 1979).
A. K. Srivastava and N. Misra (1047). Comput. Theor. Chem. 2014, 1–5.
E. Kadossov and U. Burghaus (2008). J. Phys. Chem. C 112, 7390–7400.
M. Samadizadeh, A. A. Peyghan, and S. F. Rastegar (2016). Main Group Chem. 15, 107–116.
M. Nayebzadeh, A. A. Peyghan, and H. Soleymanabadi (2014). Physica E 62, 48–54.
J. Kakemam and A. A. Peyghan (2013). Comput. Mat. Sci. 79, 352–355.
R. Shinde and M. Tayade (2014). J. Phys. Chem. C 118, 17200–17204.
Acknowledgements
A. K. Srivastava acknowledges Council of Scientific and Industrial Research (CSIR), New Delhi, India for a research fellowship [Grant No. 09/107(0359)/2012-EMR-I]. The authors acknowledge the Department of Science and Technology, Government of India, New Delhi, for providing the Computational Facilities in the Department of Chemistry, Indian Institute of Technology Kanpur, India. We thank to the reviewers for their valuable comments and suggestions for improving the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Srivastava, A.K., Misra, N. & Pandey, S.K. DFT Study on Planar (CaO) n Rings (n = 1–5) and Their Hydrogen Storage Behavior: Ca–O Versus Mg–O Clusters. J Clust Sci 29, 57–65 (2018). https://doi.org/10.1007/s10876-017-1306-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-017-1306-x