
Abstract
Magnesium transporter 1 (MAGT1) gene loss-of-function variants lead to X-linked MAGT1 deficiency with increased susceptibility to EBV infection and N-glycosylation defect (XMEN), a condition with a variety of clinical and immunological effects. In addition, MAGT1 deficiency has been classified as a congenital disorder of glycosylation (CDG) due to its unique role in glycosylation of multiple substrates including NKG2D, necessary for viral protection. Due to the predisposition for EBV, this etiology has been linked with hemophagocytic lymphohistiocytosis (HLH), however only limited literature exists. Here we present a complex case with HLH and EBV-driven classic Hodgkin lymphoma (cHL) as the presenting manifestation of underlying immune defect. However, the patient’s underlying immunodeficiency was not identified until his second recurrence of Hodgkin disease, recurrent episodes of Herpes Zoster, and after he had undergone autologous hematopoietic stem cell transplant (HSCT) for refractory Hodgkin lymphoma. This rare presentation of HLH and recurrent lymphomas without some of the classical immune deficiency manifestations of MAGT1 deficiency led us to review the literature for similar presentations and to report the evolving spectrum of disease in published literature. Our systematic review showcased that MAGT1 predisposes to multiple viruses (including EBV) and adds risk of viral-driven neoplasia. The roles of MAGT1 in the immune system and glycosylation were highlighted through the multiple organ dysfunction showcased by the previously validated Immune Deficiency and Dysregulation Activity (IDDA2.1) score and CDG-specific Nijmegen Pediatric CDG Rating Scale (NPCRS) score for the patient cohort in the systematic review.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Magnesium transporter 1 (MAGT1), a Mg2 + transporter in the endoplasmic reticulum, is crucial for maintaining intracellular magnesium balance. The clinical impact of MAGT1 deletion was first identified in a patient cohort by Li et Al., who described a unique phenotype associated with recurrent bacterial and viral infections, chronic Epstein-Barr virus (EBV) and low CD4+ T-cell numbers [1,2,3].
Further investigation into MAGT1 revealed its role in asparagine (N)-linked glycosylation as part of the Oligosaccharyltransferase (OST) complex [4, 5]. MAGT1 or its homologue TUSC3 associate with the enzymatic subunit of the OST complex, STT3B. The overlapping functions of MAGT1 and TUSC3 in glycosylation allow for compensation through increased TUSC3 expression following MAGT1 deletion [4, 6]. Since TUSC3 expression is undetectable in liver and immune cells, MAGT1 is required for the N-linked glycosylation in these tissues [7]. Ravell et al. showed a wide net of glycoproteins that are hypo-glycosylated in MAGT1-deficient patients, highlighting an expanding array of downstream effects [8]. Based on this crucial role of N-linked glycosylation, the etiology of the disease due to MAGT1 deficiency was renamed to XMEN (X-linked MAGT1 deficiency with increased susceptibility to Epstein-Barr virus infection and N-linked glycosylation defect).
These patients have a primary immunodeficiency (PID) with risk of recurrent viral (EBV, varicella-zoster, herpes simplex) and sinopulmonary bacterial infections [8, 9]. The regulation of intracellular magnesium is thought to be essential for cellular immunity against EBV, and MAGT1 deficiency impairs the magnesium influx required for T-cell activation [1, 3, 10]. In addition, the lack of glycosylation by MAGT1 leads to the decreased surface expression of natural killer group 2, member D (NKG2D), a vital immunoreceptor on natural killer (NK) and CD8+ T cells in the immunosurveillance and cytolytic control of EBV-infected cells and tumors [4, 11]. As a result, over 75% of XMEN patients have chronic EBV infections, and up to 50% may develop EBV-associated lymphoma by their third decade of life [9].
Approximately 80 documented cases of MAGT1 deficiency have broadened our comprehension of its implications beyond the immune system [1,2,3,4,5, 8, 12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. EBV is the most common infection associated with hemophagocytic lymphohistiocytosis (HLH), a syndrome of life-threatening hyperinflammation [54]. Considering the association with EBV, MAGT1 deficiency could theoretically predispose to developing EBV-driven HLH. For this reason, MAGT1 is included in targeted gene panels for primary HLH alongside other pathogenic genetic variants with EBV susceptibility [2, 12, 55]. This report presents a complex case of a patient diagnosed with MAGT1 deficiency following recurrent EBV-driven Classic Hodgkin lymphoma with concurrent HLH. The presentation with HLH necessitated a comprehensive understanding of the MAGT1 deficiency phenotype through a systematic literature review to characterize the full spectrum of this disease.
Methods
Human Subjects
Patient and healthy controls were enrolled at Children’s Healthcare of Atlanta. Blood sample preparation, whole exome sequencing analysis, and the assessment of rare single-nucleotide variants were performed based on institutional protocol. Index case and healthy controls were enrolled following informed consent, with approval by the Emory University Institutional Review Board.
Flow Cytometry and Analysis
Peripheral blood mononuclear cells were isolated by Ficoll-Paque PLUS (GE Healthcare) gradient density centrifugation and used for immunophenotyping. Flow cytometry data were acquired on BD FACSymphony A5 (BD Biosciences, Franklin Lakes, NJ) and analyzed using FlowJo software v10 (BD, Ashland, Ore). List of antibodies used for staining is found in Supplement Materials and Methods. Cell viability was measured using Live/Dead Aqua (Invitrogen). T-cell activation was defined by co-expression of HLA-DR+ and CD38+ on T-cell effector memory (TEM) CD4+ and CD8+ T cells [56].
Systematic Review
A comprehensive literature review of peer‐reviewed journal articles, case reports, and case series published between January 2011 (the initial identification of XMEN) and September 15, 2023 was conducted. The literature search was conducted using bibliographic databases, including PubMed, Web of Science, Scopus, and Google Scholar. The terms used for searching the databases included a combination of: “XMEN” and “MAGT1”.
Records were screened based on predetermined criteria detailed in Supplement Materials and Methods. The results of the screening and selection steps for our study are illustrated using the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) flow diagram established by Prisma guidelines (Fig. 1) [57].

Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) flow diagram detailing the study screening and selection process for XMEN/MAGT1 deficiency reports
Results
Index Case and Disease Course
A 12-year-old boy presented to our institution after two recurrences of EBV-driven classic Hodgkin Lymphoma (cHL). The patient's medical history is outlined in Fig. 2, A.

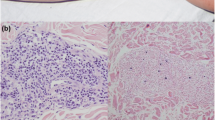
Undiagnosed MAGT1 deficiency presents as a complex, severe phenotype. (A) Timeline of patient history prior to and following presentation. Major dates in presentation and treatment course are listed along the x axis with accompanying significant events below. Above the timeline are indicated major therapies with which the patient was treated, spanning for certain periods and indicated by brackets (dark purple bar). BEACOPP: bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone. BEAM: BCNU, etoposide and cytarabine, and melphalan. (B) Top panel: Liver biopsy showing Reed-Sternberg cells (arrows) interspersed in a background of many small lymphocytes and histiocytes. H&E stain, 400X. Bottom panel: Reed-Sternberg cells are positive for EBER (red chromogen). EBV in-situ hybridization, 400X. (C) PET CT scan of patient at presentation shows metastatic spread of EBV lymphoma
At 4 years of age, he presented with fever, night sweats, weight loss, adenopathy, and splenomegaly, initially attributed to an EBV infection. Investigations revealed pancytopenia, elevated ferritin, triglycerides, and liver enzymes, with hemophagocytosis on axillary lymph node biopsy. Accordingly, he was diagnosed with HLH and commenced treatment with dexamethasone and etoposide. Following initiation of HLH therapy, review of the axillary lymph node and marrow biopsies revealed underlying stage IVB nodular sclerosing cHL with Reed-Sternberg cells positive for EBV-encoded small RNA (EBER). Bilateral bone marrow biopsies confirmed the cHL diagnosis and demonstrated hemophagocytosis.
Following stage IV cHL diagnosis, he received four cycles of BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), followed by two cycles of ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) and consolidative radiation to the right axillary lymph nodes, spleen, supraclavicular fossa and right axilla. He attained remission and, surveillance CT scans over 3 years showed no disease.
At 8 years old, he experienced intermittent fevers, fatigue, and right cervical adenopathy. Subsequent imaging and cervical lymph node biopsy confirmed cHL relapse. He was treated with three cycles of ifosfamide and vinorelbine, with follow-up imaging indicating a good response. He underwent an autologous stem cell transplant with BEAM conditioning (BCNU, etoposide and cytarabine, and melphalan) followed by 16 cycles of Brentuximab infusions, and remained in remission for an additional two and a half years. (Fig. 2A).
His past history was also significant for infectious complications. He had recurrent severe respiratory and ear infections, intermittent disseminated molluscum contagiosum, and shingles at ages four, six, and eight years. He also had persistent hypogammaglobulinemia from the age of four, but only received IVIG infusions intermittently.
Upon referral to our institution at 48 months into 2nd remission, the patient had been experiencing three months of fevers, weight loss, and fatigue. A CT scan revealed widespread lymphadenopathy, with an inguinal lymph node biopsy suspicious for an EBV-driven process. Liver biopsy and sampling of epigastric lymph nodes revealed an infiltrate with large, atypical EBER-positive Reed-Sternberg cells, confirming lymphoma recurrence (Fig. 2B). PET imaging showed widespread metastatic disease in lymph nodes, skeleton, liver, spleen, and brain parenchyma (Fig. 2C). Bilateral bone marrow biopsy revealed hemophagocytosis, raising suspicion for HLH.
Laboratory evaluation at our institution revealed lymphopenia, anemia, elevated Ferritin of 3,256 ng/mL, sIL-2R level elevated at 5,195 IU/ml, and EBV PCR elevated at 146,398 IU/mL. Additional elevated cytokines were concerning for EBV-driven HLH (Table 1). For confirmation, we assessed the expression of activation markers on CD4+ and CD8+ T-cell subsets. Compared to healthy control, there was a reduced naïve CD8+ population, however CD4:CD8 ratio was greater than 2, similar to control (Fig. 3A). Furthermore, our patient showed increased activation markers HLA-DR+ and CD38+ expression in the TEM compartment of CD4+ and CD8+ T cells, consistent with HLH (Fig. 3B).

HLH in MAGT1 deficiency is marked by increase in activation of CD4 + and CD8 + TEM cells. (A) Representative FACS plots defining CD4+ and CD8+ T cells from CD3+ T cells in healthy control and patient. T-cell phenotypes are identified from expression of CCR7 and CD45RA. Gating strategy is listed above corresponding plots. (B) Representative FACS plots showing surface expression of HLA-DR+ CD38 + markers on the TEM compartment of CD4+ and CD8+ T cells in healthy control and patient, TEM: T effector Memory, FACS: Fluorescence-activated cell sorting. (C) NKG2D expression levels in CD8+ T cells of patient is lower compared to three healthy controls. The geometric MFI is indicated beside each expression histogram. MFI: mean fluorescence intensity
Despite initiation of nivolumab, fever persisted for four days with increasing ferritin levels. Subsequently, tocilizumab was administered due to concern for persistent hyperinflammation. IL-6 blockade resulted in fever resolution and decreased ferritin.
Concurrently, he underwent genetic evaluation for an underlying immune defect contributing to his severe, atypical clinical course [PID gene panel from Invitae Corporation, 407 genes]. Results revealed a novel deletion of Exons 9–10 in the MAGT1 gene, indicative of MAGT1 deficiency. Confirmatory testing revealed markedly reduced levels of NKG2D expression in CD8+ T cells when compared to healthy controls (Fig. 3C). Additional immunological studies demonstrated markedly reduced NK cells compared to controls, decreased B cells with reduced isotype-switched CD27+IgM−IgD− memory B cells, and an increase in CD8+ effector memory cells (Table S1). Surrogate confirmatory testing was performed using the Carbohydrate Transferrin Test (CDT), which measures the degree of glycosylation in transferrin. Our patient’s CDT demonstrated an increase in mono-oligo transferrin isoforms relative to the normal di-oligo fraction, along with an abnormal apolipoprotein CIII-0/CIII-2 ratio, suggestive of a congenital disorder of glycosylation (CDG) with impaired N-glycosylation (Fig. S1).
Given the recurrence of EBV-driven lymphoma, the patient received a 3-month course of brentuximab and nivolumab, followed by a repeat PET scan revealing a favorable response. Other than immune defects, he did not have other symptoms commonly associated with CDGs like growth failure, developmental delays, or other neurologic manifestations, but did have elevated transaminases (Table S1). With the lymphoma in remission, he was evaluated for allogeneic HSCT to address the underlying immune defect.
Updated Spectrum of MAGT1 Deficiency
We performed a systematic review of MAGT1/XMEN literature which yielded 272 publications from all sources (Table S3). After removing duplicates, we retrieved 156 articles in the English literature. Articles were filtered based on relevancy. The first round of exclusions focused on titles and abstracts and the second based on a full text review. From these exclusions, 50 articles were identified and abstracted (Table S3) [1, 2, 4, 8,9,10, 13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53, 58,59,60,61]. Of these, 16 articles [2,3,4, 8,9,10, 16,17,18, 20,21,22,23,24,25, 58] were also reviewed in the most recent review by Ravell et al. [9] and we update this with 34 additional articles.
From these 50 articles, we abstracted 80 unique cases of MAGT1 deficiency (Table S4). To analyze the relevant clinical phenotypes, we applied further stringent criteria, excluding patients without disclosure of MAGT1 genetic variant (n = 7), patients with MAGT1 variant identified in large whole exome panels for HLH/Rhabdomyolysis/PID cohorts (n = 9), and patients for whom there was only a genetic variant disclosed without further documentation of phenotype (n = 2) (Table S4). One article disclosed a patient with intellectual disability (ID) and abnormal skin findings with a concomitant genetic variant in ATRX, an ID-causing gene, and thus was excluded [22]. Our focus was thus on confirmed and documented cases, which amounted to 61 patients (Table S4).
Baseline Characteristics and EBV Susceptibility
Among these patients, various pathogenic variants were identified, including missense (n = 2), splice (n = 6), frameshift (n = 22), nonsense (n = 27), and large deletions (n = 4) (Fig. 4 and Table S2). The most prevalent variant, c.991C > T, p.(Arg331Ter), occurred in 6 individuals from 5 different families across diverse regions with varying phenotypes [4, 8, 19, 45, 47]. All 61 patients were male with a median of 12 years (range 8 months – 58 years) at diagnosis. From our cohort, HSCT has been performed on 8 individuals with severe EBV-driven disease, with a high mortality of 50%. Altogether, 7/61 patients experienced mortality, four due to complications from HSCT (at age 17–45 years) and three others from uncontrolled lymphoma or associated complications (at age 5 – 58 years) (Table S4).

Schematic representation of MAGT1 protein domains and pathogenic variants seen in systematic review. Important domains of MAGT1 gene include the thioredoxin (TRX) domain and OST Complex subunit OST3/OST6. Lollipop graph based on MAGT1_Human gene (RefSeq: NM_032121) generated from cBio Portal and further modified with BioRender.com [71,72,73]. Green circles indicate missense Pathogenic variant subtypes are shown in colored circles, green for missense, orange for splice, pink for frameshift, brown for nonsense and black brackets indicating sites of large deletions. Frequency of each pathogenic variant is seen on the y axis. For large deletions, frequency is shown in parentheses
40 out of 61 patients tested positive for EBV by PCR, while 16 tested negative and 5 had unknown EBV status. MAGT1 deficiency was confirmed by low NKG2D surface expression in NK and T cells in all 53 tested individuals (Fig. 5A).

MAGT1 deficiency comprises a variability of phenotypes in multiple organ systems. (A) Clinical manifestations of MAGT1 deficiency in systematic review grouped by organ systems. The percentage of all patients who presented with the specific clinical manifestation or laboratory finding is found on the X axis. The number of patients reported with each clinical phenotype out of the number of patients tested is given for each. (B) Pie chart representation of the frequency of different neoplastic tumors in systematic review. The number of each neoplasia subtype is shown. * Non-Malignant Lymphoproliferation includes chronic lymphadenopathy/splenomegaly/hepatomegaly, reactive follicular hyperplasia and EBV-LPD
Lymphoproliferation/Neoplasia
Non-malignant lymphoproliferation was prevalent in 33 out of 57 patients, presenting in a spectrum from chronic lymphadenopathy/splenomegaly/hepatomegaly to biopsy-proven reactive follicular hyperplasia and EBV-driven lymphoproliferative diseases (LPDs) (Fig. 5A). Aggressive EBV-driven LPDs were found in six of these patients at non-lymphoid sites including skin, hypothalamus, palate, and muscle tissue and required chemotherapeutic agents for treatment. In addition, two patients also exhibited Castleman-like changes, one with HHV8-Negative Multicentric Castleman disease.
Neoplasia was diverse affecting 16 individuals (one with both cHL and Burkitt lymphoma) with initial diagnosis at 4 to 45 years of age and median of 15 years (Fig. 5A and Table S4). Predominant lymphoma types included cHL, Diffuse Large B-cell Lymphoma, Burkitt Lymphoma, and unspecified Non-Hodgkin Lymphoma (Fig. 5B). Other tumors documented were HHV8 + Kaposi Sarcoma, EBV + Marginal Zone Lymphoma, Cutaneous T-cell Lymphoma, and Liposarcoma.
Immune Deficiency
Immune deficiency characterized by recurrent ear and sinopulmonary infections was noted in 39 out of 57 patients. The presence of persistent EBV viremia, disseminated molluscum contagiosum, skin warts, mouth sores, and severe sinopulmonary infections was prevalent (Fig. 5A).
Analysis of immune profile data from 55 patients revealed markedly elevated B cells, low CD4/CD8 ratio, heightened αβ- double CD4+ and CD8+ negative T cells and CD4 lymphopenia (Fig. 5A). Notably, B-cell phenotyping indicated an increase in naïve B cells and a decrease in class-switched memory B cells. Hypogammaglobulinemia was prominent, with 32 and 33 out of 55 patients having low levels of IgG and IgA, respectively. This was consistent in patients whether or not rituximab was used for treatment. One patient had low IgM levels, and seven showed significantly reduced antibody responses, particularly to polysaccharides. 16 of the 55 patients were on immunoglobulin replacement due to recurrent infections and hypogammaglobulinemia (Fig. 5A). Patients who required rituximab as part of chemotherapy for LPD/neoplasms or hemolytic anemia received IVIG more consistently than those who did not.
Autoimmunity/Hyperinflammation
Autoimmunity affected 18 out of 55 individuals, with severe autoimmune cytopenias such as autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP) with platelets less than 25,000/uL identified in 13 patients (Fig. 5A). Two cases of refractory ITP necessitated splenectomy. Additional autoimmune manifestations included Guillain-Barré syndrome, autoimmune hepatitis, episcleritis, alopecia partialis, central nervous system vasculitis, ulcerative colitis, tenosynovitis, thrombocytopenia, and transient neutropenia. Hyperinflammation was rare, with one patient experiencing hemophagocytosis at the age of 45 due to EBV-driven lymphoma [2, 9]. Moreover, two patients were diagnosed with Kawasaki disease at the early ages of 3 and 17 months [3, 33, 46].
Utilizing the IDDA2.1 Score for MAGT1 Deficiency
We used the previously validated Immune Deficiency and Dysregulation Activity (IDDA2.1) score generated by Seidel et al. to evaluate MAGT1 deficiency in comparison to other primary immunodeficiencies with EBV susceptibility (Table S5) [62, 63]. In our study, we applied the IDDA2.1 score to assess 12 organ systems related to immune dysregulation in 55 out of 61 patients for whom a full immune workup and comprehensive organ involvement was disclosed. The Karnofsky performance score was set at 100% for standardization due to unavailability in our patient cohort. Hospitalization and ICU stay details were excluded from the IDDA2.1 score due to lack of disclosure. A heatmap of the 55 patients was generated, following Seidel et al.'s main categories (Table S5 and Fig. 6A). By organizing patients based on decreasing severity of EBV/chronic infections, a notable phenotypic distinction emerged, revealing a higher incidence of neoplasia and a more severe prognosis in EBV-positive individuals. To enhance visualization, we employed a radar chart similar to Seidel et al. illustrating that the most significant features of MAGT1 Deficiency include severe infections, hypogammaglobulinemia, other organ or immune dysfunction and lymphoproliferation (Fig. 6B).

IDDA2.1 and NPCRS Scores highlight main phenotypes of MAGT1 deficiency. (A) Phenotype expression heatmap showing results of grading according to previously described IDDA2.1 score for 17 defined parameters. The clustered heatmap was created by using the R package pheatmap 1.0.12 (Raivo Kolde, 2019). Clustering on patients was done in the order of decreasing severity of EBV chronic infection with a separation based on seropositivity. Severity of each domain is expressed as 0,1,2,3,4 according to IDDA2.1 scale corresponding to white, yellow, light orange, dark orange, red color in heatmap. This score consists of 22 parameters, each quantified per patient from 0 to 4. For more information on criteria, please refer to Supplement Table 5 and Seidel et al. publication on IDDA2.1 score [63]. (B) Representation of described 17 parameters of IDDA2.1 score as a radar (spider) chart, with values for each as percentages similar to kaleidoscope function by Seidel et al. [63]. The frequency of each portrayed complication or affected organ in the cohort corresponds to the dot in an axis from 0 to 100% [63]. (C) Representation of domain 2 of the Nijmegen Pediatric CDG Rating Scale (NPCRS) as a radar chart. This encompasses 10 categories: seizures, encephalopathy, bleeding diathesis or coagulation defect, gastrointestinal, endocrine, respiratory, cardiovascular, renal, liver function, and blood anomalies further defined in Supplement Table 5. The frequency of each portrayed complication or affected organ in the cohort corresponds to the dot in an axis from 0 to 100%
Multisystem Abnormalities
Most patients with MAGT1 deficiency exhibited non-immune organ dysfunction. Liver involvement was observed in 39 out of 61 patients, with 33 individuals showing fluctuating elevated liver enzymes and 11 displaying varying degrees of steatosis and bridging fibrosis on hepatic biopsies (Fig. 5A). Carbohydrate deficient transferrin testing in 21 patients revealed characteristics consistent with Type 1 congenital disorders of glycosylation (CDG) with elevated Di-Oligo Transferrin levels, and six of them also had abnormalities in apolipoprotein-CIII glycosylation (Fig. 5A). Seven patients experienced bleeding dysfunction including severe hemorrhage, epistaxis, hematochezia, and perioperative and non-operative bleeding episodes. Less common features included pericardial and/or pulmonary effusions and profound growth hormone deficiency.
Neurological abnormalities were present in 16 out of 56 patients, with brain MRI findings showing rare occurrences like cave septum pellucidum in five patients from four different families (Fig. 5A). Two patients experienced neurodegeneration and cognitive decline in the second decade, with imaging findings indicating cerebral, cerebellar, brainstem, and spinal atrophy, along with multifocal white matter lesions and calcification [33, 46]. Additional neurological symptoms included movement problems, lower limb weakness, and acute onset hemiplegia. Three cases exhibited characteristics more akin to classical congenital disorders of glycosylation (CDG), such as intellectual and developmental delay and mild facial dysmorphism [4]. Four patients showed chemotherapy-related consequences, including brain atrophy and posterior reversible encephalopathy syndrome (PRES).
To assess non-immune CDG dysfunction in MAGT1 deficiency, the Nijmegen Pediatric CDG Rating Scale (NPCRS) was utilized [64]. This validated tool measures disease severity in CDG disorders across three domains: current functional status, system-specific involvement, and current clinical assessment regarding growth, development, and neurologic deficits [65, 66]. Our focus was on Domain 2 of the NPCRS score, covering system-specific involvement in our cohort of 55 patients as detailed prior (Table S5). A radar chart was employed to highlight the systems most affected by glycosylation defects in MAGT1 deficiency, indicating liver involvement in 87.5% of patients, blood abnormalities in 41%, bleeding/coagulation issues in 12.5%, and other systems, including neurological, to a lesser degree (Fig. 6C). The neurological manifestations of the disease were not fully encapsulated by the NPCRS domain 2 score which primarily focuses on encephalopathy and seizures and are likely much more global with abnormal anatomic findings and signs of neurodegeneration as previously discussed.
Discussion
In this study, we present a case of MAGT1 deficiency with a severe phenotype, initially manifested by EBV-driven lymphoma with associated complication of HLH at the age of four. The lack of early genetic testing despite the severe presentation highlights the importance of a high index of suspicion in young children with EBV-driven lymphoma. Early genetic diagnosis could have facilitated initiation of supportive therapy and consideration for allogeneic HSCT as a potential curative option. While abnormal immune profile was observed throughout the patient's history, no other non-immune manifestations of MAGT1 deficiency were prominent except for elevated liver function studies and increased frequency of viral and sinopulmonary infections.
Our systematic review delved into the broad spectrum of immune effects in MAGT1 deficiency and its association with HLH. While familial HLH is commonly associated with pathogenic variants in PRF1, UNC13D, STX11, and STXBP2; some immunodeficiencies, including XLP and MAGT1 deficiency, lead to an inability to clear EBV, contributing to HLH [54, 55]. Our systematic review found one patient other than our case with HLH as part of his EBV-driven lymphoma, who unfortunately succumbed after HSCT due to hemophagocytic syndrome and multiorgan failure [2, 12]. One of the largest studies of 1892 patients with suspected genetic HLH disorders discovered five patients with MAGT1 deficiency that were referred for genetic testing following diagnosis or suspicion of HLH [32]. Interestingly, two other studies from different geographical areas also found two novel genetic variants mutations in MAGT1 following whole exome panels for patients meeting HLH criteria [29, 36]. Hence, MAGT1 has been included in whole exome and targeted sequencing panels for patients with HLH.
MAGT1 deficiency lies on the crossroads of immunodeficiency and congenital disorders of glycosylation (CDG). Lymphoproliferation is a consistent hallmark of MAGT1 deficiency, irrespective of EBV seropositivity, and encompasses a broad presentation from benign lymphoproliferative manifestations that are Castleman-like to EBV-associated LPD and classical lymphoma subtypes (including EBV-negative). Moreover, rare non-hematopoietic lineage neoplasia has been reported. The prototypical XMEN patient exhibits recurrent sinopulmonary infections and persistent EBV viremia, mirrored in our review cohort. However, a notable susceptibility to other chronic viruses, such as molluscum contagiosum, skin warts, and severe varicella-zoster virus (VZV) infections, underscores a broader immune deficiency in viral clearance. Certain viruses even precipitate neoplasms in these patients, including EBV Lymphoma and HHV8 Kaposi Sarcoma as seen in our cohort [23, 37]. In addition, two case reports, which met exclusion criteria, highlighted three individuals with Polyoma-virus-positive Merkel Cell Carcinoma and one with EBV-positive systemic T-cell lymphoma, underscoring the diverse viral-associated malignancies associated with MAGT1 deficiency [26, 27, 41]. MAGT1 deficiency also leads to impaired glycosylation of immune receptors, CD28 and CD70. Their important role in NK cell and CD8+ T-cell function to clear chronic viral infections is thus compromised [5, 7]. NKG2D activity in NK cells, crucial for defense against polyomaviruses, HPV, and gamma herpesviruses like EBV, is also mediated by MAGT1. [67,68,69].
Initially characterized by CD4+ T-cell lymphopenia and an inverted CD4:CD8 T-cell ratio, the immune abnormalities associated with MAGT1 deficiency have expanded. Most patients exhibit a B-cell differentiation and maturation defect, featuring an increased number of naive B cells and decreased class-switched memory B cells. Rowane et al. recently demonstrated that this naïve-like population has a high expression of CD5 leading to problems with B-cell maturation, previously reported in mice models [51, 70]. Decreased serum IgG and IgA, coupled with impaired responses to polysaccharide antigens, are common. One patient was initially classified to have specific polysaccharide deficiency until the underlying diagnosis of MAGT1 deficiency was made [51]. MAGT1 deficiency, responsible for glycosylating IgG, results in a hypoglycosylated form with reduced half-life, explaining the vulnerability to infections [5]. Consequently, immunoglobulin replacement becomes essential for patients, including our case.
As evidenced in our index case and previous reports of EBV-driven lymphoma in MAGT1 deficient patients, it is important that recurrence of EBV-driven lymphoma in a male with or without underlying immune deficiency should warrant genetic evaluation for MAGT1 and other inborn errors of immunity with predisposition to EBV. Early management with allogenic HSCT has been shown to prevent relapse of EBV-driven lymphoma in MAGT1 deficient patients and correct the underlying immune deficiency [17,18,19]. However, due to the high mortality with HSCT, this consideration needs to be made judiciously especially if patients have MAGT1-related bleeding dysfunction [17].
The IDDA2.1 score application enabled a comprehensive assessment of MAGT1 deficiency's impact across 12 organ systems regarding immune dysregulation in our patients [61]. Our scores aligned with Seidel et al.'s CD27 and CD70 deficiency charts, with notable distinctions in heightened "other organ involvement" in our analysis [60, 61, 71]. Interestingly, CD70 has also been shown to be hypoglycosylated in MAGT1 deficiency and is thought to contribute to the lack of EBV protection, further supporting our findings [8]. The downstream effects, as evidenced in our CDG-specific NPCRS and immune specific IDDA2.1 radar charts, underscore the significance of MAGT1 deficiency in glycoprotein hypoglycosylation, leading to liver damage, bleeding dysfunction, hematological anomalies, and neurological manifestations.
Hepatocellular injury, observed in 30% of known CDG disorders, is a prominent feature of MAGT1 deficiency [72]. The spectrum of liver damage, from elevated transaminases to fibrosis and cirrhosis, remains underrecognized. Ding et al. demonstrated that TUSC3, MAGT1's homologue, exhibits the lowest expression in liver and bone marrow, emphasizing these affected uncompensated systems [13]. Bleeding dysfunction, prevalent in other CDGs, results from glycosylation alterations affecting coagulation balance. MAGT1 deficiency, implicated in platelet dysfunction via glycosylation defects in targets like PAR-1, leads to dysregulated calcium signaling in platelets [22, 57]. Studies by Gotru et al. revealed heightened platelet aggregation and degranulation in MAGT1-deficient platelets [48]. In addition, Kauskot et al. showcased that HSCT reversed the predisposition to bleeding through corrected platelet progenitors [19]. The intricate interplay of glycoproteins in platelets determines the bleeding or thrombosis tendency observed in our cohort. Neurological abnormalities and midline defects, noted in other CDGs linked to PMM2 and ALG6, are also evident in MAGT1 deficiency [74]. The pronounced neurodegeneration necessitates further research to unravel MAGT1's role in these manifestations.
Our study has limitations, including the retrospective nature of obtaining patient data from literature and reliance on reported symptoms rather than primary analysis of patient records. As well, due to limited phenotype details, certain patients with MAGT1 pathogenic variants met exclusion criteria in our analysis. Grading for IDDA2.1 and NPCRS scores is subject to some variation due to missing values and interpretation in scoring domains.
MAGT1 deficiency manifests in diverse phenotypes, involving immune system anomalies and multi-organ dysfunction. Our patient case highlights the complexity of XMEN syndrome, underscoring the need for comprehensive genetic and immunologic evaluation in patients with recurrent infections and hematologic malignancies. HLH may be an underrecognized manifestation of MAGT1 deficiency, potentially more common than previously thought. The broad impact of glycosylation in various organ systems, specifically the immune system, in MAGT1 deficiency remains incompletely understood.
Data Availability
Relevant data/methods utilized in the preparation of this manuscript are provided in the manuscript and electronic supplementary material.
Abbreviations
- HLH:
-
Hemophagocytic lymphohistiocytosis
- EBV:
-
Epstein–Barr virus
- XMEN:
-
X-linked magnesium deficiency with Epstein-Barr virus (EBV) infection and neoplasia
- PID:
-
Primary Immunodeficiency
- cHL:
-
Classic Hodgkin Lymphoma
- CDG:
-
Congenital Disorder of Glycosylation
- HSCT:
-
Hematopoietic stem cell transplant
- IVIG:
-
Intravenous Immunoglobulin
- sIL-2R:
-
Soluble interleukin-2 receptor
- IFN-γ:
-
Interferon gamma
- IDDA2.1:
-
Immune Deficiency and Dysregulation Activity Score
- ID:
-
Intellectual Disability
- NPCRS :
-
Nijmegen Pediatric CDG Rating Scale
References
Li F-Y, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature. 2011;475:471–6.
Li F-Y, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein-Barr virus. Blood. 2014;123:2148–52.
Li F-Y, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, et al. Signaling role for Mg2+ revealed by immunodeficiency due to loss of MagT1. Nature. 2011;475:471–6.
Blommaert E, Péanne R, Cherepanova NA, Rymen D, Rymen D, Staels F, et al. Mutations in MAGT1 lead to a glycosylation disorder with a variable phenotype. Proc Natl Acad Sci USA. 2019;116:9865–70.
Matsuda-Lennikov M, Biancalana M, Zou J, Zou J, Ravell JC, Zheng L, et al. Magnesium transporter 1 (MAGT1) deficiency causes selective defects in N-linked glycosylation and expression of immune-response genes. J Biol Chem. 2019;294:13638–56.
Cherepanova NA, Shrimal S, Gilmore R. Oxidoreductase activity is necessary for N-glycosylation of cysteine-proximal acceptor sites in glycoproteins. J Cell Biol. 2014;206:525–39.
Ding H, Li Y, Fang M, Chen J, Liu L, Lu Z, et al. Epigenetic activation of the TUSC3 gene as a potential therapy for XMEN disease. J Allergy Clin Immunol. 2023;151:1622-1633.e10.
Ravell JC, Matsuda-Lennikov M, Chauvin SD, Zou J, Zou J, Biancalana M, et al. Defective glycosylation and multisystem abnormalities characterize the primary immunodeficiency XMEN disease. J Clin Inv. 2019;130:507–22.
Ravell JC, Chauvin SD, He T, Lenardo M. An Update on XMEN Disease. J Clin Immunol. 2020;40:671–81.
Chaigne-Delalande B, Li F-Y, O’Connor GM, Lukacs MJ, Jiang P, Jiang P, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science. 2013;341:186–91.
Shabani M, Nichols KE, Rezaei N. Primary immunodeficiencies associated with EBV-Induced lymphoproliferative disorders. Crit Rev Oncol Hematol. 2016;108:109–27.
Cohen JI, Jaffe ES, Dale JK, Pittaluga S, Heslop HE, Rooney CM, et al. Characterization and treatment of chronic active Epstein-Barr virus disease: a 28-year experience in the United States. Blood. 2011;117:5835–49.
Chauvin SD, Chauvin SD, Price S, Zou J, Hunsberger S, Brofferio A, et al. A double-blind, placebo-controlled, crossover study of magnesium supplementation in patients with XMEN disease. J Clin Immunol. 2021;42(1):108–18.
Iyengar VV, Chougule A, Gowri V, et al. XMEN saved by magnesium. Scand J Immunol. 2022;95:e13154.
Brault J, Meis RJ, Li L, Bello E, Liu T, Sweeney CL, et al. MAGT1 messenger RNA-corrected autologous T and natural killer cells for potential cell therapy in X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection and neoplasia disease. Cytotherapy. 2020;23:203–10.
Akar HH, Patıroglu T, Hershfield M, van der Burg M. Combined immunodeficiencies: twenty years experience from a single center in Turkey. Cent Eur J Immunol. 2016;41:107–15.
Dimitrova D, Rose JJ, Uzel G, Cohen JI, Rao KV, Bleesing JJ, et al. Successful Bone Marrow Transplantation for XMEN: Hemorrhagic Risk Uncovered. J Clin Immunol. 2019;39:1–3.
Klinken E, Gray PA, Pillay B, Worley L, Edwards ESJ, Edwards ESJ, et al. Diversity of XMEN Disease: Description of 2 Novel Variants and Analysis of the Lymphocyte Phenotype. J Clin Immunol. 2020;40:299–309.
Kauskot A, Mallebranche C, Bruneel A, Fenaille F, Solarz J, Viellard T, et al. MAGT1 deficiency in XMEN disease is associated with severe platelet dysfunction and impaired platelet glycoprotein N-glycosylation. J Thromb Haemost. 2023;21:3268–78.
Dhalla F, Sarah MS, Sadler R, Chaigne-Delalande B, Sadaoka T, Soilleux E, et al. Identification of a novel mutation in MAGT1 and progressive multifocal leucoencephalopathy in a 58-year-old man with XMEN disease. J Clin Immunol. 2015;35:112–8.
Patiroglu T, Akar HH, Akar HH, Gilmour K, Unal E, Ozdemir MA, et al. A case of XMEN syndrome presented with severe auto-immune disorders mimicking autoimmune lymphoproliferative disease. Clin Immunol. 2015;159:58–62.
Qiao Y, Mondal K, Trapani V, Wen J, Carpenter G, Wildin R, et al. Variant ATRX Syndrome with Dysfunction of ATRX and MAGT1 Genes. Hum Mutat. 2014;35:58–62.
Brigida I, Chiriaco M, Di Cesare S, Cittaro D, Di Matteo G, Giannelli S, et al. Large deletion of MAGT1 gene in a patient with classic kaposi sarcoma, CD4 lymphopenia, and EBV infection. J Clin Immunol. 2017;37:32–5.
He TY, Xia Y, Li CG, Li CR, Qi ZX. [X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection, and neoplasia: report of a family and literature review]. Yang J Zhonghua Er Ke Za Zhi. 2018;56:48–52.
Hoyos-Bachiloglu R, Concha S, Sepúlveda P, Campos R, Perez-Mateluna G, King A, et al. The many faces of XMEN disease, report of two patients with novel mutations. J Clin Immunol. 2020;40:415–7.
Mohsin N, Jabbour AJ, Nghiem P. 33904 Early onset Merkel cell carcinoma in patients with XMEN disease. J Amer Acad Dermatol. 2022;87:47.
Moshin N, Hunt D, Jabbour AJ, Nghiem P, Freemam AF, Bergerson JRE, et al. Early-onset merkel cell carcinoma is associated with germline defects in DNA repair genes and a rare immunodeficiency. J Investig Dermatol. 2022;142:2839.
Reynolds C, Stein C, Atkinson T, Hurst A, Kimberlin D. P267 XMEN disease: an unexpected presentation of a rare primary immunodeficiency. Ann Allergy Asthma Immunol. 2017;119:S67–8.
Mukda E, Trachoo O, Pasomsub E, Tiyasirichokchai R, Iemwimangsa N, Sosothikul D, et al. Exome sequencing for simultaneous mutation screening in children with hemophagocytic lymphohistiocytosis. Int J Hematol. 2017;106:282–90.
Verdugo CA, King AV. XMEN: MAGT1 Mutation Associated Immunodeficency. Case report of an atypical presentation. J Clin Immunol. 2019;39:S33–S33.
Freeman C, Bauer C, Miller H, Wright B, Rukasin C, Badia P. M278 XMEN disease: an unexpected presentation with an unexampled mutation. Ann Allergy Asthma Immunol. 2020;125:S92–3.
Gadoury-Levesque V, Dong L, Su R, Chen J, Zhang K, Risma KA, et al. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv. 2020;4:2578–94.
Ochoa S, Toro C, Wolfe L, Durkee-Shock J, Lenardo M, Binder K, et al. Expanding The Phenotype Of XMEN disease: MAGT1 deficiency presenting with neurodegenerative symptoms. J Clin Immunol. 2020;40:S76–7.
Kruijt N, van den Bersselaar LR, Kamsteeg EJ, Verbeeck W, Snoeck MMJ, Everaerd DS, et al. The etiology of rhabdomyolysis: an interaction between genetic susceptibility and external triggers. Eur J Neurol. 2021;28:647–59.
Jalil M, Rowane M, Rajan J, Hostoffer R. Successful Anti-SARS-CoV-2 Spike Protein Antibody Response to Vaccination in MAGT1 Deficiency. Allergy Rhinol. 2021;12:21526567211056240.
Bąbol-Pokora K, Wołowiec M, Popko K, Jaworowska A, Bryceson YT, Tesi B, et al. Molecular Genetics Diversity of Primary Hemophagocytic Lymphohistiocytosis among Polish Pediatric Patients. Arch Immunol Ther Exp. 2021;69:31.
Freeman CM, Freeman C, Wright BL, Bauer CS, Rukasin CR, Chiang SCC, et al. Cutaneous T-cell lymphoma as a unique presenting malignancy in X-linked magnesium defect with EBV infection and neoplasia (XMEN) disease. Clin Immunol. 2021;226:108722–108722.
Serrano-Santiago S, Sosa-Castellanos N, Nazario S, Mujca AA. XMEN disease: MAGT1 mutation in a child with hypogammaglobulinemia. Ann Allergy Asthma Immunol. 2022;129:S144–5.
Huang X, Liu D, Gao Z, et al. Case report: EBV-positive extra-nodal marginal zone lymphoma associated with XMEN disease caused by a novel hemizygous mutation in MAGT1. Front Oncol. 2021;11:653266–653266.
Haskologlu S, Baskin K, Aytekin C, Islamoglu C, Ceylaner S, Dogu F, et al. Scales of Magt1 gene: novel mutations, different presentations. Iran J Allergy Asthma Immunol. 2022;21:92–7.
Man J, Cao P, Wang H, Qian X, Miao H, Zhu X, et al. 089 - Report of systemic ebv-positive t-cell lymphoma of childhood associated with XMEN disease caused by a novel mutation. Leuk Res. 2022;121:S55.
Guha S, Khetrapal P. Recurrent oral ulcers due to XMEN syndrome. 2022;51:139.
Au EY, Tung EKK, Ki Ip RW, Li PH. Novel MAGT1 mutation found in the first chinese XMEN in Hong Kong. Case Rep Immunol. 2022;2022:1–3.
Watson CM, Nadat F, Ahmed S, Crinnion LA, O’Riordan S, Carter C, et al. Identification of a novel MAGT1 mutation supports a diagnosis of XMEN disease. Genes Immun. 2022;23:66–72.
Peng X, Lu Y, Wang H, Wu B, Gan M, Xu S, et al. Further delineation of the spectrum of XMEN disease in six Chinese pediatric patients. Front Genet 2022;13:768000.
Ebrahim A, Benavides D, Leonardo M, Gahl W, Wolfe L, Ravell J, et al. Adult-onset neurodegeneration in XMEN (P11-9.002). Neurology. 2022;98.
Chen S, Wang X, Sun C, Zhao C-B, Lin J. MAGT1 gene mutation is associated with myositis and CD127 expression downregulation. J Clin Immunol. 2023;43:315–8.
Gotru SK, Mammadova-Bach E, Sogkas G, Schuhmann MK, Schmitt K, Kraft P, et al. MAGT1 deficiency dysregulates platelet cation homeostasis and accelerates arterial thrombosis and ischemic stroke in mice. Arterioscler Thromb Vasc Biol. 2023;43:1494–509.
de Gaulmyn M, Guery R, Néel A, Masseau A, Agard C, Fieschi C, et al. Déficit immunitaire en MAGT-1: à propos d’un cas. Rev Med Interne. 2023;44:A196.
Nielsen C, Nilsson C, Assing K, Herlin MK, Skakkebæk A, Larsen M, et al. Compromised PAR1 activation—A cause for bleeding in XMEN? Thromb Haemost. 2023;123:641–4.
Rowane MJ, Stewart-Bates BC, Doll RJ, Meyerson HJ, Venglarcik JS, Callahan M, et al. CD5 B-Cell predominant primary immunodeficiency: part of the spectrum of MAGT1 deficiency. Ther Adv Allergy Rhinol. 2023;14:27534030231199676.
Schutt M, Gunderman L, Lippner E, Ahmed A, Khanolkar A, Khojah A. An unexpected diagnosis of MAGT1 deficiency in a patient with CVID-like features. Molluscum Contagiosum Atopy Clin Immunol. 2023;250:109454.
Mohanty MC, Taur PD, Sawant UP, Yadav RM, Potdar V. Prolonged fecal shedding of SARS-CoV-2 in asymptomatic children with inborn errors of immunity. J Clin Immunol. 2021;41:1748–53.
Marsh RA. Epstein-barr virus and hemophagocytic lymphohistiocytosis. Front Immunol. 2018;8:1902.
Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020;135:1332–43.
Chaturvedi V, Marsh RA, Zoref-Lorenz A, Owsley E, Chaturvedi V, Nguyen TC, et al. T-cell activation profiles distinguish hemophagocytic lymphohistiocytosis and early sepsis. Blood. 2021;137:2337–46.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
Li F-Y, Lenardo MJ, Chaigne-Delalande B. Loss of MAGT1 abrogates a Mg2+ flux required for T cell signaling and leads to a novel human primary immunodeficiency. Magnes Res. 2011;24:S109–14.
Li F-Y, Chaigne-Delalande B, O’Connor G, Lukacs M, Zheng L, Shatzer A, et al. Intracellular free Mg2+ is required to maintain NKG2D expression necessary for controlling EBV infection in XMEN disease (P3028). J Immunol. 2013;190:114.15–114.15.
Agharahimi A, Ravell J, Sun A, Bergerson J, Rosenzweig S, Rao VK, et al. Spectrum of malignancy in MAGT1 deficiency (XMEN). J Clin Immunol. 2020;40:S99-100.
Blommaert E, Cherepanova NA, Staels F, Wilson MP, Gilmore R, Schrijvers R, et al. Lack of NKG2D in MAGT1-deficient patients is caused by hypoglycosylation. Hum Genet. 2022;141:1279–86.
Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol. 2020;145:1452–63.
Seidel MG, Tesch VK, Yang L, Hauck F, Horn AL, Smolle MA, et al. The immune deficiency and dysregulation activity (IDDA2.1 ‘Kaleidoscope’) score and other clinical measures in inborn errors of immunity. J Clin Immunol. 2022;42:484–98.
Achouitar S, Mohamed M, Gardeitchik T, Wortmann SB, Sykut-Cegielska J, Ensenauer R, et al. Nijmegen paediatric CDG rating scale: a novel tool to assess disease progression. J Inherit Metab Dis. 2011;34:923–7.
Ligezka AN, Mohamed A, Pascoal C, Ferreira VDR, Boyer S, Lam C, et al. Patient-reported outcomes and quality of life in PMM2-CDG. Mol Genet Metab. 2022;136:145–51.
Witters P, Andersson H, Jaeken J, Tseng L, van Karnebeek CDM, Lefeber DJ, et al. D-galactose supplementation in individuals with PMM2-CDG: results of a multicenter, open label, prospective pilot clinical trial. Orphanet J Rare Dis. 2021;16:138.
Della Chiesa M, De Maria A, Muccio L, Bozzano F, Sivori S, Moretta L. Human NK cells and herpesviruses: mechanisms of recognition, response and adaptation. Front Microbiol. 2019;10.
Mishra R, Welsh RM, Szomolanyi-Tsuda E. NK Cells and virus-related cancers. Crit Rev Oncog. 2014;19:107–19.
Mishra R, Chen AT, Welsh RM, Szomolanyi-Tsuda E. NK cells and γδ T cells mediate resistance to polyomavirus-induced tumors. PLoS Pathog. 2010;6:e1000924.
Gotru SK, Gil-Pulido J, Beyersdorf N, Diefenbach A, Becker IC, Vögtle T, et al. Cutting edge: imbalanced cation homeostasis in MAGT1-deficient B cells dysregulates B cell development and signaling in mice. J Immunol. 2018;200:2529–34.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
de Bruijn I, Kundra R, Mastrogiacomo B, Tran TN, Sikina L, Mazor T, et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR project GENIE biopharma collaborative in cBioPortal. Cancer Res. 2023;83:3861–7.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Funding
This work was supported by NHLBI 1K08HL141635-01A1, Atlanta Pediatric Scholars Program K12 Scholar supported by grant K12HD072245 and U54AI082973 (to S.C.). Emory + Children's (Pediatric) Flow Cytometry Core facility was used for flow cytometry studies.
Author information
Authors and Affiliations
Contributions
K.G. was involved in data collection and analysis, creation of figures and manuscript writing. W.M., S.M., S.P., S.C. and A.A. were involved in manuscript writing. D.K. was involved in data analysis, and manuscript writing. S.C. was responsible for project concept, oversight and manuscript writing. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Disclosure of Conflicts of Interest
S.C. serves on the advisory committee for SOBI. The rest of the authors declare no relevant conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Golloshi, K., Mitchell, W., Kumar, D. et al. HLH and Recurrent EBV Lymphoma as the presenting manifestation of MAGT1 Deficiency: A Systematic Review of the Expanding Disease Spectrum. J Clin Immunol 44, 153 (2024). https://doi.org/10.1007/s10875-024-01749-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10875-024-01749-y