
Abstract
Two cadmium(II) complexes based on 2-(3-(pyridin-2-yl)-1H-pyrazol-1-yl)benzoate with chemical formulas of [Cd2(ppb)4(H2O)2]·8H2O (1) and [Cd(Hppb)I2]n (2) (Hppb = 2-(3-(pyridin-2-yl)-1H-pyrazol-1-yl)benzoic acid), have been synthesized. Both complexes are well characterized by IR, TGA and X-ray single-crystal as well as powder diffraction. Structural analyses reveal that 1 is a binuclear complex and 2 has one-dimensional polymers, respectively, in which they are expanded to three-dimensional supramolecular structures by hydrogen bonds. The Cd(II) coordinated environments in 1 are slightly distorted octahedral geometries, but central atoms in 2 display distorted square pyramidal geometries. There are obvious differences in ligation modes of the ppb− or Hppb ligands for both complexes. In 1, four ppb− ligands are divided into two groups, displaying µ1-kN, N′ and µ2-kN, N′: kO coordination modes. Two ppb− ligands act as two µ1,6-bridges linking the binuclear Cd(II) cations with a distance of 6.282(2) Å. In 2 the neutral Hppb ligands exhibit a µ2-kN, N′: kO coordination mode, and connecting Cd(II) ions by µ1,6-bridges and forming an infinite 1D chain along b axis. Hirshfeld surfaces and fingerprint plots analyses indicate that weak interactions of C–H···π, π···π, O–H···O or O–H···I play important roles in crystal packings of 1 and 2. Luminescent measurements in solid-state show their different intensities in the luminescences of 1 and 2.
Graphical Abstract
A binuclear complex [Cd2(ppb)4(H2O)2]·8H2O (1) and a one-dimensional complex [Cd(Hppb)I2]n (2) were synthesized and characterized by X-ray crystal diffraction. Investigation of solid-state luminescence properties indicates the blue shifts of the emission λmax of complexes.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Much attention has been paid to the construction of metal complexes due to the various structural patterns and their tremendous potential applications [1, 2]. Generally, the constructions of metal complexes are dependent on a variety of factors such as the organic ligands, solvents, metal ions, ratio of reactants, and counter ions [3, 4]. The incorporation of the counter ion within the charged framework is one of the strategies followed in the synthesis of metal complexes and this approach is employed for different reasons. For example, the counter ion can act as templates, co-ligands or they can modulate the synthesis mediae. In this way, the anion templates have been used in the synthesis of different kinds of complexes. The anion may be needed to balance the charge request, such as F−, Cl−, Br−, I−, N3−, SCN−, SO42−, etc. [5]. Thus, besides organic ligands and metal centers, counter ions (Including their the type, size, shape and geometry) may be important components in the assembly of metal coordination complexes. The anions act as not only the guests or templates in the construction of host frameworks, but also the auxiliary ligands in monodentate or multidentate modes to coordinate to the metal centers, which tune the coordination numbers and geometry of the metal centers, further affecting the physical and chemical properties of these complexes [6].
2-(3-(Pyridin-2-yl)-1H-pyrazol-1-yl)benzoic acid (Hppb) contains one carboxylate and two nitrogen heterocyclic groups with versatile coordination positions [7]. Considering the special d10 electronic configuration of cadmium(II) ion, it is selected as a metal ion to get novel metal complexes with luminescent properties [8,9,10,11]. In this text, cadmium(II) complexes [Cd2(ppb)4(H2O)2]·8H2O (1) and [Cd(Hppb)I2]n (2) were successfully received. The as-synthesized samples were characterized by single-crystal and powder X-ray diffraction, elemental analysis, TGA and infrared spectra. Moreover, their luminescent properties were also studied.
Experimental
All chemicals were of analytical reagent grade and used without further purification.
Physical Measurements
The IR spectra for 1 and 2 were recorded in the range of 4000–400 cm−1 on a Bruker TENSOR27 spectrometer as KBr pellets. Elemental analyses were carried out using a CHNO Rapid instrument. Powder X-ray diffraction (PXRD) data were collected on a Bruker D8 Advance with Cu Kα radiation (λ = 1.5418 Å). Thermogravimetric analyses (TGA) were carried out with a Dupont thermal analyzer in the temperature range of 25–800 °C under N2 flow with a heating rate of 5 °C min−1. Fluorescence spectra in solid were characterized at room temperature by a Varian-Cray Eclipse fluorescence spectrophotometer.
X-ray Crystallography and Structure Solution
Single-crystal X-ray diffraction data of complex 1 were collected in the beamline 3W1A of the Beijing Synchrotron Radiation Facility (BSRF) at 100 K with a MARCCD-165 detector (λ = 0.7200 Å) with the storage ring working at 2.5 GeV. Data were processed by using HKL 2000. The DISP instructions were used in the SHELXL2018 refinements in order to correct anomalous scattering values (f′ and f″) of elements for the synchrotron wavelength used. The diffraction data of complex 2 were collected on a Bruker Smart ApexII diffractometer equipped with a 1 K CCD instrument by using a graphite monochromator utilizing Mo-Kα radiation (λ = 0.71073 Å) at room temperature.
Crystal data, data collection and structure refinement details are summarized in Table 1. H atoms attached to C atoms were placed geometrically and refined using a riding model approximation, with C–H = 0.93 Å and Uiso(H) = 1.2 Ueq(C). H atoms bonded to O atoms were located firstly in a difference Fourier map and were refined isotropically, with the isotropic displacement parameters coupled to the anisotropic displacement parameters (ADPs) of the parent O atoms, Uiso(H) = 1.5 Ueq(O). Tentative free refinements of their positional coordinates resulted in an unsatisfactory wide range of O–H distances; thus, O–H bond lengths were therefore restrained to 0.82(1) Å. The O–H distances were located in the range of 0.820–0.835 Å in 1 and 0.83 Å in 2. Crystallographic data of the complexes 1 and 2 were shown in Table 1. Selected bond lengths and bond angles were given in Table 2.
Hirshfeld Surface
Hirshfeld surfaces [12, 13] and related graphical tools [14, 15] have been shown to enhance exploration of the nature of the interactions between molecules in crystals. The Hirshfeld surface partitions crystal space into smooth non-overlapping volumes associated with each molecule, and is defined implicitly where the ratio of promolecule to procrystal electron densities equals 0.5. Since the local nature of the surface is dictated by the electron density and position of neighboring atoms inside and outside the surface, it reflects in considerable detail the immediate environment of a molecule in a crystal, and summarizes all intermolecular interactions in a remarkable graphical fashion [16].
Preparation of the Complexes
Synthesis of [Cd2(ppb)4(H2O)2]·8H2O (1)
To a solution of Hppb (26.5 mg, 0.10 mmol) in 5 mL of distilled water in a 50 mL of flask, added dropwise a Cd(NO3)2·4H2O (30.9 mg, 0.10 mmol) in 5 mL of distilled water with constant stirring, and then 0.20 mol/L KOH (0.50 mL) was added. The mixture solution was stirred for 8 h at RT. The resulting solution was filtered and the filtrate was kept for slow evaporation at room temperature. After a few days, colorless block-shaped crystals of 1 with a yield of 6.4 mg (based on Cd). Anal. Calcd.(%) for 1 (C60H60Cd2N12O18, MW 1462.02): C 49.29, H 4.14, N 11.50%. Found: C 49.23, H 4.15, N 11.52%. IR (cm−1): 3410(m), 3119(m), 1609(s), 1586(m), 1438(w), 1391(s), 1159(m), 1118(m), 1098(m), 976(m), 951(w), 771(s), 697(s), 635(m), 464(w), 420(w).
Synthesis of [Cd(Hppb)I2]n (2)
A mixture of Cd(NO3)2·4H2O (30.9 mg, 0.10 mmol), Hppb (26.5 mg, 0.10 mmol), NaI (15.0 mg, 0.10 mmol), 0.20 mol/L KOH (0.50 mL) and 10 mL H2O was heated at 433 K for 72 h under autogenous pressure in a sealed 25 mL of Teflon-lined stainless steel vessel. Colorless crystals suitable for X-ray diffraction were obtained with a yield of 7.1 mg (based on Cd). Anal. Calcd.(%) for 2 (C15H11CdN3O2I2, MW 642.8): C 28.53, H 1.76, N 6.65%. Found: C 28.58, H 1.72, N 6.68%. IR (cm−1): 1606(s), 1572(m), 1506(s), 1432(m), 1366(s), 1154(m), 1093(m), 972(m), 949(w), 799(m), 761(s), 498(m), 473(w), 402(w).
Results and Discussion
Synthesis, Characterization and FT-IR Spectra
Ligand Hppb undergoes two types of in situ reactions as shown in Scheme 1, resulted in complexes 1 and 2 exhibiting a binuclear and a 1D linear chain structures, respectively. Namely, complex 1 was obtained by reactions of cadmium(II) nitrate and Hppb with a molar ratio of 1:1 at room temperature, while complex 2 was obtained by reactions of cadmium(II) nitrate, Hppb and sodium iodide with a molar ratio of 1:1:1 at 433 K.

Strategy for the synthesis of 1 and 2
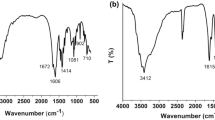
The FT-IR spectra of complexes 1 and 2 exhibit the peaks at 1609 cm−1 and 1608 cm−1, respectively, which are due to ν(CO) of coordinated carboxylate groups, demonstrating carboxyl groups of the Hppb ligand coordinated [17]. The strong characteristic bands of the coordinated carboxylate groups appear at 1586 cm−1 and 1572 cm−1 for the asymmetric stretching vibrations, and 1391 cm−1 and 1366 cm−1 for the symmetric one [18] in 1 and 2, respectively. The broad band at about 3410 cm−1 for 1 corresponds to the O–H stretching vibrations of water molecules [19].
Crystal Structure Descriptions
Crystal Structure of [Cd2(ppb)4(H2O)2]·8H2O (1)
X-ray diffraction analysis reveals that complex 1 crystallizes in the monoclinic space group C2/c. In asymmetric unit of complex 1, there are one Cd(II) cation, two ppb− anions, one coordinated water and four lattice water molecules. The whole molecule generating by a twofold axis of symmetry is a binuclear structure.
The Cd(II) atom is hexa-coordinated and displays a slightly distorted octahedral geometry. In the [CdN4O2] moiety, four N atoms in cis-N,N-chelating fashion from two deprotonated ppb− ligands and one carboxylic O atom in bridge mode from one ppb− ligand and one O atom from a coordinated water molecule with four equatorial atoms (N1i, N4, N5 and O1) and two axial atoms (O5 and N2i, symmetry code: (i) − x + 1, y, − z+ 1/2). The O5 atom is from a terminal coordinated water molecule, and the remainder atoms are from the three ppb− ligands. In 1, four ppb− ligands are divided into two groups, displaying µ1-kN, N′ and µ2-kN, N′: kO coordination modes. Two ppb− ligands act as two µ1,6-bridges linking the binuclear Cd(II) cations with a distance of 6.282(2) Å. The Cd–O coordination distances range from 2.294 (2) to 2.302 (2) Å and the Cd–N bond distances range from 2.312 (2) to 2.396 (2) Å, which are comparable to those reported for other Cd(II) compounds [20,21,22,23,24]. As shown in Fig. 1a, each carboxylic O atom from ppb− ligand in bridge modes to link adjacent Cd(II) ions to form the binuclear [Cd2(ppb)4(H2O)2].

a The binuclear structure [Cd2(ppb)4(H2O)2] of 1 (H atoms are omitted for clarity). Perspective view showing coordination environments of the Cd(II) atoms in complex 1 (30% thermal ellipsoids). Symmetry code: (i) − x + 1, y, − z + 1/2. b Perspective view of the 2D network in 1
Inter-molecular hydrogen bonds (Table 3) play an important role in stabilizing the structure. The lattice water O6 atom acts as a hydrogen-bond donor bonded to carboxyl O4 atom, viz. O6–H···O4ii, the lattice water O7 atom acts as a hydrogen-bond donor bonded to lattice water O9 atom, viz. O7–H···O9ii (Symmetry code: (ii) − x, y, − z + 1/2). As shown in Fig. 1b, the crystal structure gives rise to an infinite subloop chain with O7–H···O9ii, O9–H···O4 and O3–H···O7 hydrogen bonds in the [100] direction. These contribute to the formation of a one-dimensional hydrogen bonded polymer with a linear conformation along a axis. As shown in Fig. 1b, these 1D chains are further expanded into 2D networks through hydrogen bonds involving O9–H···O8iii (symmetry code: (iii) x, − y + 1, z + 1/2). In addition, inter-molecular weak interactions C–H···O and C–H···π exist in the structure (Table 3). As a result, a 3D supra-molecular network is finally built by inter-molecular interactions.
Crystal Structure of [Cd(Hppb)I2]n (2)
X-ray diffraction analysis reveals that complex 2 crystallizes in the triclinic space group PI and forms an infinite 1D chain. The asymmetric unit of 2 consists of one crystallographically independent Cd(II) center, one Hppb ligand, and two coordinated iodide ions.
The Cd(II) atom is penta-coordinated and displays a distorted square pyramidal geometry (τ = 0.40). In the [CdON2I2] moiety, two N atoms in cis-N,N-chelating fashion from one Hppb ligand and two coordinated iodide ions and one carboxylic O atom in bridge mode from one Hppb ligand, four atoms (N1, N2, I2 and O1i, (i) − x + 1, y + 1/2, − z + 1/2) occupy the basal plane, and one atoms (I1) sit on the axial position (Fig. 2a). Five-coordinated compounds usually adopt trigonal bipyramidal (TP), square pyramidal (SP), or inter-mediate coordination geometries. The angular structural parameter τ = |β − α|/60 (τ = 0 for ideal SP and 1 for ideal TP), where α and β are the first and second largest coordination angles, has been reported as a quantitative tool to determine the extent to which five-coordination geometries are more TP or SP [25]. The τ value for coordination around Cd1 is 0.40, confirming coordination geometry of Cd1 being TP (Fig. 2b).The Cd–N coordination distances range from 2.337(2) to 2.401(2) Å and the Cd–O bond distances range is 2.497(2) Å and the Cd–I bond distances range from 2.713(1) to 2.752(1) Å, which are comparable to those reported for other Cd(II) compounds [20,21,22,23,24]. The neutral Hppb ligands exhibit a µ2-kN, N′: kO coordination mode and connect Cd(II) ions by µ1,6-bridges with the distance of 8.075(4) Å, which makes the complex 2 forming an infinite 1D chain along b axis (Fig. 2c). Complex 2 shows a 2D supra-molecular structure assembled through π···π packing interactions with a distance of 3.885 (2) Å (Fig. 2d). Moreover, in the structures of 2, intermolecular hydrogen bonds and weak interactions (Table 3) play important roles in stabilizing the structure (Fig. 2e). As a result, a 3D supra-molecular network is finally built by inter-molecular interactions.

a Perspective view showing coordination environment of the Cd(II) atom in complex 2 (30% thermal ellipsoids). b The distorted square pyramidal coordination geometry of Cd(II), symmetry code: (i) x, y − 1, z. c Perspective view of the 1D chain along the b axis. d Supramolecular 2D network of 2 by π···π packing interaction. e Intermolecular hydrogen bond O2–H···I2ii in 2, symmetry code: (ii) x − 1, y + 1, z
Coordination Modes of Hppb
As shown in Scheme 2, In both complexes, Hppb exhibits three types of coordination modes: (a) anionic µ2-kN, N′:kO, (b) anionic µ1-kN, N′ and (c) neutral µ2-kN, N′:kO. Namely, Hppb display bridging modes (a) and (b) in 1, and only (c) in 2.

The coordination fashions of ligands observed in complexes 1 and 2
Hirshfeld Surface Analysis
The Hirshfeld surfaces and 2D fingerprint plots [26] of complexes 1 and 2 are illustrated in Figs. 3, 4, 5 and 6. As shown in Figs. 3 and 5, there are O–H···O (3) H-bond, C–H···π (1) and π···π (2) weak interactons in complex 1 and O–H···I (4) H-bond and π···π (2) weak interactons in complex 2. Using the CrystalExplorer software, the percentages of contacts contributed to the total Hirshfeld surface area of molecules are shown in the Figs. 4 and 6 for complexes 1 and 2 respectively. The proportions of C–H···π, π···π and O–H···O interactions are 24.5%, 5.7% and 17.4% of the total Hirshfeld surfaces for 1. However, the proportions of π···π and O–H···I interactions are 5.8%, and 28.8% of the total Hirshfeld surfaces for 2. That is, in 2, the O–H···I hydrogen bonds instead of the O–H···O interactions of 1, may be attributing to the influence of the coordination of the iodide atoms.

Hirshfeld surfaces of the complex 1 mapped with dnorm property, the molecules in tube/licorice representation within the transparent surface maps, C–H···π (1) π···π (2) and O–H···O (3)

Fingerprint plots of the complex 1: C–H···π (1) π···π (2) and O–H···O (3), listing the percentages of contacts contributed to the total Hirshfeld surface area of molecules

Hirshfeld surfaces of the complex 2 mapped with dnorm property, the molecules in tube/licorice representation within the transparent surface maps, C–H···π (1), π···π (2) and O–H···I (4)

Fingerprint plots of the complex 2: π···π (2) and O–H···I (4), listing the percentages of contacts contributed to the total Hirshfeld surface area of molecules
XRD Analysis
The as-synthesized samples of 1 and 2 are characterized by powder X-ray diffraction (XRD). As shown in Fig. 7a and b, the XRD patterns are almost consistent with the simulated spectra, demonstrating the high phase-purity of the bulk samples of the complexes.

Powder X-ray diffraction patterns of complexes 1a and 2b
TG Analyses
TGA curve of 1 exhibited two main weight loss steps (Fig. 8a). The first weight loss of 9.42% from 25 to 145 °C corresponded to the loss of eight lattice water molecules (calcd 9.85%). The second weight loss from 230 °C, the complex began to lose weight rapidly, which indicates that the structure of complex 1 begins to collapse. The decomposition for 2 occurs starting at 325 °C (Fig. 8b). The single weight loss step corresponds to the release of organic ligands and I− ions. Obviously, the thermostability of 2 is more stable than one of 1.

TGA curves for complexes 1a and 2b
Luminescence Property
The luminescent properties of complexes 1, 2 and free ligand were examined in the solid state at room temperature (Fig. 9). The luminescent properties of complexes 1, 2 and free ligand were examined in the solid state at room temperature (Fig. 9). Free Hppb, complexes 1 and 2 exhibited emissions at λmax = 379, 360, 351 nm with the corresponding excitation wavelengths of 320, 322, 280 nm, respectively.

Solid-state luminescence spectra of 1, 2 and Hppb at room temperature
In contrast to luminescent of the ligand, the emission λmax of complexes 1 and 2 displays the blue shift values of 19 and 28 nm. The observed blue-shifted of the complexes 1 and 2 may be assigned to the intra-ligand transitions (π–π*) fluorescent emission when coordinated with the metal. The coordination modes that the pyridine ring and pyrazole ring are almost coplanar boosting the electronic delocalization in the ligand′s backbone and charge transfer to the metal center, and thus greatly affect the energy gap between π* and π molecule orbitals of Hppb, and finally results in blue shift [27]. The investigation indicates that the differences between the fluorescent properties for 1 and 2 should be attributed to the different structures of them.
Concluding Remarks
Two complexes based on 2-(3-(pyridin-2-yl)-1H-pyrazol-1-yl)benzoate have been constructed successfully. The ligand Hppb exhibits different coordination modes to connect Cd(II) centres to form discrete binuclear (1) or 1D (2) structure. Hirshfeld surface analyses indicate effective roles of C–H···π, π···π,O–H···O or O–H···I contacts in crystal packings of 1 and 2. The complex 1, consists of [Cd2(ppb)4(H2O)2] through the carboxylate groups of the Hppb ligands generating a 0D binuclear unit. The complex 2, a 1D mononuclear unit with iodine countra-ion coordinated with Cd(II) ion, then the oxygen atom further bridge Cd(II) ions forming a 1D chain. The negative ions play important roles in governing the molecular structures. Halide anions are easily coordinated with Cd(II) ions than nitrate anions, and due to different structures the complexes indicates different luminescence.
Supplementary Material
The cif files of complexes 1 and 2 were deposited with the Cambridge Crystallographic Data Center (CCDC 1561701 and 1561702). The data can be obtained free of charge from authors or the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK; FAX: +44-1223-336-033; E-mail: deposit@ccdc.cam.ac.uk.
References
Moulton B, Zaworotko MJ (2001) Chem Rev 101:1629–1658
Lu W, Huo J, Feng Y, Zhao S, You H (2016) Dalton Trans 45:9676–9683
Ma LF, Wang LY, Lu DH, Batten SR, Wang JG (2009) Cryst Growth Des 9:1741–1749
Wei X, Di D, Chu W, Zhu Q, Huang R (2008) Inorg Chim Acta 361:1819–1826
Liang G, Liu Y, Zhang X, Yi Z (2014) CrystEngComm 16:9896–9906
Campos-Fernandez CS, Schottel BL, Chifotides HT, Bera JK, Bacsa J, Koomen JM, Russell DH, Dunbar KR (2005) J Am Chem Soc 127:12909–12923
Yang LB, Wang HC, Fang XD, Chen SJ, Xu QQ, Zhu AX, Yang Z (2016) CrystEngComm 18:130–142
Li RF, Liu XF, Wang YF, Feng X, Ma LF (2016) Chin J Struct Chem 35:1936–1943
Rachuri Y, Bisht KK, Parmar B, Suresh E (2015) J Solid State Chem 223:23–31
Song XZ, Song SY, Zhao SN, Hao ZM, Zhu M, Meng X, Zhang HJ (2013) Dalton Trans 42:8183–8187
Wang K, Huang XK, Zhu L, Chen ZL, Liang FP (2016) Chin J Struct Chem 35:1912–1919
McKinnon JJ, Mitchell AS, Spackman MA (1998) Chem-Eur J 4:2136
Spackman MA, Byrom PG (1997) Chem Phys Lett 267:215
McKinnon JJ, Spackman MA, Mitchell AS (2004) Acta Cryst B 60:627
Spackman MA, McKinnon JJ (2002) CrystEngComm 4:378
Spackman MA, McKinnon JJ, Jayatilaka D (2008) CrystEngComm 10:377
Gu X, Xue D (2006) Cryst Growth Des 6:2551–2557
Karmakar A, Goldberg I (2010) CrystEngComm 12:4095
Fang RQ, Zhang XM (2006) Inorg Chem 45:4801–4810
Chen X, He S, Chen F, Feng Y (2014) CrystEngComm 16:8706–8709
Yamamoto E, Kubo K, Kato N, Mori A (2000) Acta CrystallogrSect C 56:E329–E330
Murugavel R, Karambelkar VV, Anantharaman G, Walawalkar MG (2000) Inorg Chem 39:1381–1390
Xie L, Lu LP, Zhu ML (2016) Chin J Struct Chem 35:1606–1614
Avecilla F, Esteban D, Platas-Iglesias C, De Blas A, Rodriguez-Blas T (2003) Acta CrystallogrSect C 59:M93–M94
Addison AW, Rao TN (1984) J Chem Soc, Dalton Trans 1349:1356
Feng SS, Qin SD, Feng GQ, Zhu ML (2012) J Chem Crystallogr 42:621–627
Zheng SL, Yang JH, Yu XL, Chen XM, Wong WT (2004) Inorg Chem 43:830–838
Acknowledgements
The authors thank Dr Zeng-Qiang Gao at line 3W1A of BSRF for his help with the single-crystal X-ray diffraction data collection and reduction, and acknowledge the financial support by the Natural Science Foundation of China (Grant Nos 21571118 and 21671124). A portion of this work was performed on the Scientific Instrument Center of Shanxi University of China.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, LY., Lu, LP. & Zhu, ML. Two Cadmium(II) Complexes Constructed by 2-(3-(Pyridin-2-yl)-1H-pyrazol-1-yl)benzoate: Crystal Structures, Luminescent Properties and Hirshfeld Surface Analyses. J Chem Crystallogr 50, 122–132 (2020). https://doi.org/10.1007/s10870-019-00781-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-019-00781-w