
Abstract
ABCC6 is a human ATP binding cassette (ABC) transporter of the plasma membrane associated with Pseudoxanthoma elasticum (PXE), an autosomal recessive disease characterized by ectopic calcification of elastic fibers in dermal, ocular and vascular tissues. Similar to other ABC transporters, ABCC6 encloses the core structure of four domains: two transmembrane domains (TMDs) and two nucleotide binding domains (NBDs) but also an additional N-terminal extension, including a transmembrane domain (TMD0) and a cytosolic loop (L0), which is only found in some members of ABCC subfamily, and for which the function remains to be established. To investigate the functional roles of this N-terminal region, we generated several domain deletion constructs of ABCC6, expressed in HEK293 and polarized LLC-PK1 cells. ABCC6 lacking TMD0 displayed full transport activity as the wild type protein. Unlike the wild type protein, ABCC6 without L0 was not targeted to the basolateral membrane. Moreover, homology modeling of L0 suggests that it forms an ATPase regulatory domain. Furthermore, we show that the expression of ABCC6 is linked to a cellular influx of Ca2+. The results suggest that TMD0 is not required for transport function and that L0 maintains ABCC6 in a targeting-competent state for the basolateral membrane and might be involved in regulating the NBDs. These findings shed new light on a possible physiological function of ABCC6 and may explain some of the hallmarks of the clinical features associated with PXE that could contribute to the identification of novel pharmacological targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
ABCC6 belongs to the subfamily C of ATP-binding cassette (ABC) transporter superfamily. These proteins are active pumps that use the energy provided by the hydrolysis of ATP to transport a variety of biological molecules across a membrane. They are implicated in drug and antibiotic resistance, signal transduction, protein secretion, and antigen presentation (Higgins 1992). The ABCC6 gene maps to chromosome 16p13.1, consists of 31 exons and encodes a transporter protein of 1503 amino acids, also known as Multidrug Resistance-associated Protein 6 (MRP6). ABCC6 is predominantly expressed at the basolateral side of liver and kidney (Uitto et al. 2001; Matsuzaki et al. 2005). Glutathione conjugates have been identified as substrates of the ABCC6 transporter in vitro, suggesting that ABCC6 is a transporter of organic anions (Belinsky et al. 2002). Mutations in the ABCC6 gene cause Pseudoxanthoma elasticum (PXE), an autosomal recessive disorder characterized by progressive ectopic mineralization of the skin, eyes, and arteries, for which no effective treatment exists. The precise prevalence of this disease is estimated to 1:25,000–100:000, with slight prevalence of females (Chassaing et al. 2005; Finger et al. 2009). It has been suggested that PXE is a metabolic disorder, caused by the absence of an unknown factor in the blood stream, which directly or indirectly control mineralization of dermal, ocular and cardiovascular tissues (Uitto et al. 2001). Recent studies have shown that the presence of ABCC6 in HEK293 cells results in the excretion of ATP and other nucleoside triphosphates, which are rapidly converted into nucleoside monophosphates and PPi, of which the latter is an inhibitor of mineralization, by an ENPP-type ectonucleotidase (Jansen et al. 2013).
Although there is no 3D structure of ABCC6 (Ostuni et al. 2011), its sequence shows a common structural organization found in all ABC transporters, which consists of a canonical “MDR-like core region” composed of two transmembrane domains (TMD1 and TMD2), each with six membrane-spanning helices and two nucleotide-binding domains (NBD1 and NBD2) which harbor conserved sequence motifs that bind ATP (ter Beek et al. 2014). ABCC6 contains an additional N-terminal transmembrane domain (TMD0) with five membrane-spanning helices (Cuviello et al. 2015) connected to the MDR-like core with a cytoplasmic linker region (L0) (Slot et al. 2011). L0 is not only found in the seven human ABCC subfamily members with TMD0 but also in the N-terminal portion before the MDR-like core in the remaining five ABCC transporters lacking TMD0. It can be assumed that the presence of these regions adds functional features to the ABCCs with respect to other human ABC transporters that lack this region. Whereas the possible roles of the additional N-terminal region in localization and transport activity have been extensively studied in other ABCCs (Bakos et al. 2000; Mason and Michaelis 2002; Westlake et al. 2005; Winkler et al. 2012), nothing is known about this region in ABCC6. In this study, we have investigated if TMD0 and L0 of ABCC6 contribute to transport function and intracellular targeting.
Materials and methods
Generation of ABCC6 variants
The following deletion mutants were created based on protein domain predictions of the sequence of ABCC6 (residue numbers corresponding to uniprot: O95255): ΔTMD0L0 (295–1503), ΔTMD0 (195–1503), TMD0 (1–194) and TMD0L0 (1–305). The ABCC6 variants were generated by mutagenesis PCR using pcDNA3flag/ABCC6 recombinant vector, which was subcloned from pcDNA 3.1D/V5-His-TOPO (Armentano et al. 2008) as a template and the QuikChange II XL Site-Directed kit (Stratagene), according to the manufacturer’s instructions. Oligonucleotide primers used are shown in ESM 1. All constructs were verified by sequencing (Eurofins MWG Operon, Ebersberg, DE) and expressed with an N-terminal flag-tag (MDYKDDDDK).
Cell culture and transfections
Both human hepatocellular carcinoma cell line (HepG2), human embryonic kidney (HEK) 293 and polarized Pig Kidney Epithelial cells (LLC-PK1) cells were maintained in Dulbecco’s modified Eagle’s medium (with 4.5 g/L glucose (Euroclone), 10 % fetal bovine serum (Euroclone), 2 mM L-glutamine (Sigma), 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma) at 37 °C in a humidified incubator with 5 % CO2. One day before transfection cells were seeded in 12-well plates at the density of 2 x 105 cells per well. Transient transfection was performed with Lipofectamine (Invitrogen) according to the manufacturer’s instructions. Stable LLC-PK1 cell clones were selected in G418 (500 μg/ml) for 10 days. To inhibit the N-linked glycosylation of proteins, transfected HEK293 cells were grown in medium containing 3 μg/ml tunicamycin.
Stably ABCC6 silenced HepG2 cells were obtained as previosly described (Miglionico et al. 2014).
Immunoblotting
Forty-eight hours after transfection, HEK293 cells were lysed in Laemmli sample buffer (60 mM Tris–HCl, pH 6.8, 10 % glycerol, 2 % SDS, 1 % β-mercaptoethanol and 0.002 % bromophenol blue) and sonicated for 1 min on ice. The lysates were heated for 5 min at 90 °C, separated on 8 or 15 % SDS-PAGE gels and transferred to PVDF membranes (Amersham Bioscience). Membranes were blocked for one hour in TBST (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 0.05 % Tween 20) with 5 % non-fat milk and incubated overnight at 4 °C with monoclonal anti flag antibody M5 (dilution 1:1000; Sigma). Membranes were washed in TBST and incubated for one hour with horseradish peroxidase–conjugated anti-mouse secondary antibody (dilution 1:10,000; Sigma). Reactive proteins were revealed with SuperSignal West Pico Chemiluminescent Substrate detection system (Pierce, Rockford, IL) and Chemidoc XRS detection system equipped with Image Lab Software for image acquisition (Biorad). Tubulin was used to normalize the amount of proteins.
Calcein efflux assay
After 48 h of transfection with pcDNA3flag, ABCC6, ΔTMD0L0 and ΔTMD0, HEK293 cells were incubated with 1 μM calcein-AM in PBS at 37 °C for 30 min, washed and incubated with 200 μl of PBS for 15, 30, 45 and 60 min. Calcein efflux was quantified spectrofluorimetrically using Glomax multi detection system (Promega) and normalized to μg of total protein.
Immunofluorescence
Stable clones of LLC-PK1 were grown on semipermeable filters, fixed in cold methanol for 10 min and blocked in saturation buffer (1 % bovine serum albumin in PBS) for 20 min at room temperature before incubation with monoclonal anti flag antibody M5 diluted 1:200 in saturation buffer for 2 h at RT. After 3 washes in PBS, cells were incubated with 1:1000 diluted Alexa 488-conjugated anti-mouse IgG (Cell Signaling) in saturation buffer for 1 h at RT. Following three washes, nuclei were stained with 1.5 μM propidium iodide in PBS. Confocal images were obtained with a laser scanning fluorescence microscope Leica TSC-SP2 (HCX PL APO, ×63/1.32–0.60 oil objective); 8-bit images were saved at 1024 × 256 and acquired using the Leica Confocal Software W.
Structural homology model of L0
A homology model of L0 of ABCC6 was created with Modeller (Fiser and Sali 2003).
Intracellular Ca 2+ measurements
ABCC6-transfected HEK293 cells were seeded on poly-L-lysine-coated glass coverslips (Ø 40 mm) and loaded with 5 μM Rhod-2 (Lifetechnologies) for 20 min at 37 °C in DMEM. HepG2 and HepG2 ABCC6-KO cells were seeded on glass coverslips (Ø 25 mm) and loaded with 6 μM Fura-2 (Lifetechnologies) for 30 min at 37 °C in DMEM. Cells were perfused with Ringer’s Solution (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM Hepes pH 7.4, 5 mM Glucose, 1.0 mM CaCl2) and stimulated with a variety of drugs as described in the results including ATP, cyclopiazonic acid and ionomycin (all from Sigma-Aldrich, St. Louis, USA). For fluorescence measurements, the coverslips with dye-loaded cells were mounted in a perfusion chamber (FCS2 Closed Chamber System, BIOPTECHS, Butler, U.S.A.) and measurements were performed using an inverted microscope (Nikon Eclipse TE2000-S microscope) equipped for single cell fluorescence measurements and imaging analysis. The sample was illuminated through a 40X oil immersion objective (NA = 1.30). The Rhod-2-loaded sample was excited at 550 nm every 5 s. The Fura-2 loaded sample was excited alternately at 340 and 380 nm every 5 s. Emitted fluorescence was passed through a dichroic mirror, filtered respectively at >600 nm or 510 nm (Omega Optical, Brattleboro, VT, USA) and captured by a cooled CCD camera (CoolSNAP HQ, Photometrics, Tucson, AZ, USA). Fluorescence measurements were performed using Metafluor software (Molecular Devices, MDS Analytical Technologies, Toronto, Canada). For determination of intracellular Ca2+ concentration images were corrected for the background fluorescence and calibrated as described by the manufacturer (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Results and discussion
To gain more insight into the functional role of the N-terminal region of ABCC6, we have characterized deletion mutants of ABCC6 lacking TMD0 and L0, and performed in silico analysis of L0 leading to findings that suggest a new role of ABCC6.
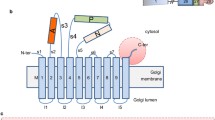
Two N-terminally flag-tagged ABCC6 constructs were generated to analyze the role of the N-terminal region of ABCC6 in transport activity: a variant lacking TMD0 (ΔTMD0) and a variant lacking both TMD0 and L0 (ΔTMD0L0) (Fig. 1). As ABCC6 full length, they were expressed in HEK293 cells. As shown in Fig. 2a, the ABCC6 protein variants were expressed at comparable level to that of wild type ABCC6. We performed transport assays tracking calcein efflux mediated by wild type ABCC6 and the variant versions, according to a previously described experimental setup (Hegedus et al. 2002; Boraldi et al. 2003). As shown in Fig. 2b, the calcein efflux was about two-fold higher in ABCC6-transfected HEK293 compared to mock cells, demonstrating a significant ABCC6-mediated calcein efflux over the background. ΔTMD0 showed a transport activity comparable to that of ABCC6 wild type, while ΔTMD0L0 was similar to that of mock cells and consistent with a loss of the transport activity in the absence of L0. To verify if ΔTMD0L0 was localized at the plasma membrane, we analyzed its polarized trafficking in stably-transfected polarized LLC-PK1 cells by immunofluorescence confocal microscope analysis (Fig. 3a). Interestingly, while ΔTMD0 was targeted to the basolateral side of the plasma membrane such as the full-length ABCC6, ΔTMD0L0 was trapped in intracellular compartments, implicating that L0 is necessary for the correct delivery of ABCC6 to the basolateral membrane. Taken together, these findings suggest that TMD0 is not required for ABCC6 transport function and that L0 is clearly involved in the basolateral membrane localization.

Schematic structures of full-length ABCC6 and its truncated versions. ABCC6 contains the typical MDR-like core region, which includes two nucleotide binding domains (NBD1 and NBD2), two transmembrane domains (TMD1 and TMD2) and an additional amino-terminal transmembrane domain (TMD0) connected to the core domain through a cytosolic linker L0. The following deletion mutants were created ΔTMD0L0 (295–1503), ΔTMD0 (195–1503), TMD0 (1–194) and TMD0L0 (1–305)

Expression and activity of ABCC6 and its N-terminal truncated versions. (a) Analyses of full length ABCC6 and N-terminal truncated variants expression in HEK293 cells by Western blotting. Total cell lysates from transiently transfected HEK293 cells were subjected to electrophoresis and immunoblot analysis with anti-FLAG antibody. Tubulin was used to normalize the amount of proteins. (b) Calcein efflux assay in HEK293 cells transfected with full length ABCC6 and N-terminal truncated variants. ΔTMD0 protein is still functional, while ΔTMD0L0 lost the transport activity. Results are expressed as mean percentages of calcein release ± SE for 3 independent experiments. **P < 0.01, Student t-test for unpaired data

Cellular localization of ABCC6 and its truncated variants in polarized LLC-PK1 monolayers by confocal laser scanning microscopy. The localization of ABCC6 and its variants were detected by indirect immunofluorescence (green signal) with anti-FLAG antibody. The upper part of each panel shows the planar XY view of a confocal plane crossing the nucleus of the monolayer, and the lower part shows a vertical XZ projection. Nucleic acids were detected by counterstaining with propidium iodide (red signal). (a) ABCC6 and ΔTMD0 are selectively expressed at the basolateral membrane (green signal), while ΔTMD0L0 shows intracellular localization (green signal). (b) TMD0L0 is selectively expressed at the basolateral membrane, while TMD0 shows intracellular localization. (c) Immunoblot analyses of C-terminal truncated variants of ABCC6 in HEK293 cells. Total cell lysates from HEK293 cells transiently expressing the indicated constructs and grown in the presence or absence of tunicamycin were subjected to electrophoresis and immunoblot analysis with anti-FLAG antibody. Tubulin was used to normalize the amount of proteins. TMD0L0 is fully glycosylated, while TMD0 is partially glycosylated
To evaluate if L0 contains a sorting signal sufficient per se for targeting ABCC6 to the basolateral membrane, we made C-terminal truncation constructs and expressed the resulting TMD0 and TMD0L0 in HEK293 cells. The cellular localization of TMD0 and TMD0L0 was analyzed by expression in LLC-PK1 cells. Confocal microscope analysis showed that TMD0L0 was predominantly targeted to the basolateral membrane of the cells and that TMD0 alone was completely retained intracellularly (Fig. 3b). The presence of a small fraction of TMD0L0 localized to intracellular vesicles may further suggest that regions downstream of L0 impose a conformational status of the L0 sequence for maximal basolateral membrane targeting, as for ΔTMD0 (Fig. 3a). As shown by Western Blotting (Fig. 3c), while TMD0L0 is completely glycosylated, a major fraction of TMD0 is not glycosylated. Since the two constructs are identical until residue 194 (including the glycosylation acceptor asparagine in position 15), they should have been equally recognized by the co-translational N-linked glycosylation machinery. Asparagine 15 has been shown to be N-glycosylated before with ABCC6 expressed in MDCKII cells (Sinkó et al. 2003) and with the first 102 residues of MRP6 expressed in vitro (Ostuni et al. 2013) and in HEK-293 T cells (Lee et al. 2014). The different levels of glycosylation observed here between TMD0 and TMD0L0 suggest that TMD0 has been deglycosylated by the ER quality control system. Thus, these findings suggest that L0 not only contains a basolateral sorting signal but that L0 contributes to folding ABCC6 into a cellular sorting-competent state, which is necessary to pass the ER quality control system and continue through the secretory pathway.
Our conclusions for the role of L0 in folding of ABCC6 are consistent with the suggested role of L0 of human MRP1, which is also localized at the basolateral membrane, based on its expression in insect Sf9 and polarized MDCKII cells with various deletion and chimeric constructs (Bakos et al. 1998; Bakos et al. 2000). Similar experiments on apical membrane-localized of human MRP2 showed that its TMD0 is necessary for correct targeting, which was most evident by the co-expression of TMD0L0 with the MDR-like core that restored full glycosylation, correct routing and transport. Therefore, our results are also not in contradiction to those reporting that a PDZ-like domain in the very C-terminus of ABCC6 has a role in membrane insertion and/or cellular targeting (Xue et al. 2014).
To find further evidence for the proposed role of L0 in ABCC6 we performed sequence and structure analysis. A conserved sequence of approximately 60 residues can be found within the about 100 amino acid long L0 in all ABCC subfamily members (ESM 1). This conserved L0 domain was found to be distantly related to a domain found in the bacterial protein ScpB with Swiss Model (Biasini et al. 2014), despite very low sequence identity. In bacteria, ScpB forms a complex with ScpA that regulates the ATPase activity of Structure-Maintenance-of-Chromosome (SMC) proteins (Volkov et al. 2003). A structural homology model of the conserved L0 domain of ABCC6 was created (ESM 1) on the basis of an ScpB structure (Kamada et al. 2013), which suggests that this domain is formed by three α-helices followed by a few β-strands. The model of the L0 domain of ABCC6 is consistent with the conserved positions occupied by hydrophobic core residues and solvent accessible charged residues (ESM 1). The N-terminal helix (in blue) of the conserved L0 domain of ABCC6 corresponds to the part of ScpB that interacts with ScpA (ESM 1). These findings suggest that L0 of ABCCs could have a role in interacting directly or through an intermediate protein with the NBDs for the regulation of their ATPase activity and consequently transport function apart from being important for the correct routing. This hypothesis is supported by the findings in MRP1 (Bakos et al. 2000) and Ycf1p (Mason and Michaelis 2002), for which the former co-expression of L0 with the MRD1-like core restored transport activity and similar deletion mutants in Ycf1p had the same effect. The first helix of ABCC6 L0 also corresponds to the previously predicted amphipathic α-helix of L0 of MRP1 and Ycf1p, which has been suggested to interact with the MDR-like core and to be required for transport activity but not for targeting (Bakos et al. 2000; Mason and Michaelis 2002). Furthermore, the transport activity of Ycf1p is regulated by a phosphorylation of residue S251 (Paumi et al. 2008), which is consistent with the solvent-exposed position of its corresponding residue (S244) between helix 2 and 3 in ABCC6 L0 homology model. Based on these considerations, it is therefore very likely that L0 of ABCC6 is involved in regulation of NBD ATPase activity directly or via a binding partner.
We also found that a small region of ScpA, the binding partner of ScpB, shares sequence homology with a region enclosing transmembrane helix S3 of members of the short Transient Receptor Potential Channel family (TRPC, ESM 1), which consist of a total of six transmembrane segments of which the last two form a tetrameric Ca2+ channel pore. Support for the idea that ABCC6 could interact with an ion channel comes from an analogous system where the ABCC6 homologues ABCC8 and ABCC9 (sulfonylurea-binding receptors SUR1 and SUR2, respectively) form a large membrane protein complex with ATP-sensitive potassium channels (Kir6) (Mikhailov et al. 2005). In ABCC8, L0 has been suggested to interact with the N-terminal cytosolic tail of Kir6 (Babenko and Bryan 2002) and to be a part of a glibenclamide binding site (Mikhailov et al. 2001). In both ABCC8 and ABCC9, L0 together with TMD0 are involved in trafficking and gating of Kir6s (Chan et al. 2003; Fang et al. 2006). The formation of the SUR-Kir6 complex has been suggested to mask ER retention signals in both proteins allowing only the complex to be exported to the plasma membrane.
TRPC channels have been postulated to be the Ca2+ entry membrane pathway after store depletion. They may behave as receptor-operated Ca2+ entry (ROCE), when they are activated by a Gq-coupled GPCR pathway mediated by PLC. Members of this family may also behave as store-operated Ca2+ entry (SOCE), when they are activated directly in response to depletion of intracellular Ca2+ stores in ER (for review see Birnbaumer 2009).
To investigate the possible connection between ABCC6 and a TRPC channel, we measured Ca2+ influx in HEK293 cells overexpressing GFP-ABCC6 in response to ATP, which acting through purinergic receptors may both activate a PLC mediate pathway and deplete Ca2+ stores. In these experiments positive cells were distinguished from controls by the typical green fluorescence emitted upon proper illumination. Figure 4a shows that exposure to 100 μM ATP elicited a cytosolic Ca2+ transient that was significantly larger in ABCC6-expressing HEK293 cells loaded with Rhod-2 when compared with wild type cells. The expression of ABCC6 did not affect ATP-induced intracellular Ca2+ release from the ER, as demonstrated by using the Ca2+-selective ionophore ionomycin, which deplete ER Ca2+ in the absence of extracellular Ca2+ (Fig. 4a, right inset). Thus, these results suggest that the significant additional increase recorded in the ATP-mediated Ca2+ response is related to ABCC6-dependent activation of Ca2+ channels at the plasma membrane. In addition, we analyzed the role of ABCC6 in the ATP-induced Ca2+ influx using HepG2 cells, a suitable in vitro model system for the study of human hepatocytes in which ABCC6 protein is endogenously expressed (de Boussac et al. 2010). We repeated the response to 100 μM ATP in Fura-2 loaded wild type HepG2 cells and previously characterized ABCC6 knockdown HepG2 cells (Miglionico et al. 2014). As shown in Fig. 4b, the ATP-induced increase in [Ca2+]i recorded in wild type HepG2 cells (black trace) was significantly attenuated in ABCC6-KO HepG2 cells (green trace). No differences in the resting [Ca2+]i were observed between HepG2-ABCC6-KO and -control cells (data not shown).

Real time evaluation of ATP-induced [Ca2+]i changes. (a) Real time evaluation of ATP-induced [Ca2+]i changes. Rhod-2 loaded HEK293 cells either expressing ABCC6 (red trace, representative of n = 75 cells) or not (black trace, representative of n = 55 cells) were challenged with the Ca2+ mediated agonist ATP. Histogram showing the ATP-induced Δ[Ca2+]i in wild type- (black bar, Δ[Ca2+]i: 90.61 ± 13.30; n = 70 cells) and ABCC6- (red bar, Δ[Ca2+]i: 158.92 ± 12.36; n = 96 cells, P < 0.01) HEK293 cells. Data are expressed as means ± SE. (**P < 0.01 vs ABCC6; Student t-test for unpaired data). Histogram showing the effect of exposure to 5 μM ionomycin in the absence of extracellular Ca2+ in HEK293 wild type (black bar, Δ[Ca2+]i: 513.08 ± 19.55; n = 65 cells) and HEK293-ABCC6 (red bar, Δ[Ca2+]i: 499.56 ± 20.76; n = 95 cells, P = n.s.) cells. Data are expressed as means ± SE. (b) Real time evaluation of ATP-induced [Ca2+]i changes in Fura-2 loaded HepG2 wild type (black trace, representative of n = 67) or ABCC6-knockout (green trace, representative of n = 72) cells. Right inset. Histogram showing the ATP-induced Δ[Ca2+]i in wild type (black bar, Δ[Ca2+]i: 133.10 ± 13.49; n = 67 cells) and ABCC6-KO (green bar, Δ[Ca2+]i: 98.04 ± 6.59; n = 72 cells, P < 0.01) HepG2 cells. Data are expressed as means ± SE. (*P < 0.05 vs ABCC6-KO; Student t-test for unpaired data). (c) Time course of changes of [Ca2+]i before, during, and after application of 20 μM CPA in the absence or presence of extracellular Ca2+ (2.0 mM) in Fura-2 loaded HepG2 wild type (black trace, representative of n = 59) or ABCC6-knockout (green trace, representative of n = 60) cells. Right inset. Summarized data of the amplitude of CPA-induced ER Ca2+ release in the absence of extracellular Ca2+ (black bar, Δ[Ca2+]i: 157.00 ± 17.50; n = 59 cells; green bar, Δ[Ca2+]i: 173.28 ± 22.4; n = 60 cells, P = n.s.), and amplitude of CPA-induced capacitative Ca2+ entry (CCE) (black bar, Δ[Ca2+]i: 275.11 ± 26.7; green bar, Δ[Ca2+]i: 180.05 ± 30.5, P < 0.01). HepG2 wild type, black bars; HepG2-ABCC6 KO, green bars. Data are expressed as means ± SE. (**P < 0.01 vs ABCC6-KO; Student t-test for unpaired data)
We next sought to specifically investigate whether the increased Ca2+ influx triggered by ATP in wild type HepG2 cells was related to an ABCC6-dependent activation of a Ca2+ entry pathway at the plasma membrane. In Fura-2 loaded HepG2 wild type cells (black trace), the inhibition of the sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) by 20 μM cyclopiazonic acid (CPA) in the absence of extracellular Ca2+ evoked a transient cytosolic Ca2+ increase (Fig. 4c) due to release of Ca2+ from the stores. Restoration of extracellular [Ca2+] to 2.0 mM in the continuous presence of CPA evoked a second cytosolic [Ca2+] increase of larger magnitude. The latter increase in cytosolic [Ca2+] represents the Ca2+-entry through the plasma membrane. Interestingly, the absence of ABCC6 (green trace) did not affect the Ca2+ increase evoked by CPA in the absence of extracellular Ca2+ while Ca2+ entry elicited in presence of 2.0 mM extracellular Ca2+ was significantly reduced (Fig. 4c).
All together these results suggest that the impaired response to ATP in the absence of ABCC6 is not due to an effect on the intracellular Ca2+ stores or basal Ca2+ level rather suggesting the lack of activation of an ABCC6-modulated Ca2+ channel at the plasma membrane. The dynamics of Ca2+entry, modulated by the presence/absence of ABCC6 in both HEK293 and HepG2 cells, is typical of that mediated by TRP channels’ family, including TRCP channels.
As suggested by the data in ESM 1, it is indeed possible that ABCC6 forms a complex with a TRPC Ca2+ channel through interactions mediated by L0 between the NBDs of ABCC6 and the ion channel as part in regulating its gating (as suggested by Fig. 4 and ESM 1), and in the formation of this complex, ER retention signals could be masked (as suggested by Fig. 3). Moreover, these findings imply that the L0s of all ABCCs may function in conjunction to ion channels.
However, we cannot exclude that other ROCE or SOCE, such as Transient Receptor Potential Vanilloid (TRPV) and Transient Receptor Potential Melastatin (TRPM) Ca2+ channels might be involved in the ABCC6 Ca2+ entry in the liver.
Although future studies are warranted for the molecular identification of ABCC6 interaction partners and how these components interact in a complete mechanism, this is the first time in which data suggest that ABCC6 is linked to cellular influx of Ca2+ and that this influx might be stimulated by ATP. Dysfunctional ABCC6 could contribute to ectopic mineralization by both decreased cellular ATP release and reduced cellular entry of extracellular Ca2+. Hopefully, this insight will be useful for the scientific community for new projects aiming at development of novel therapeutic strategies in the treatment of PXE.
Abbreviations
- ABC:
-
ATP binding cassette
- ER:
-
endoplasmic reticulum
- NBD:
-
nucleotide binding domain
- PPi:
-
pyrophosphate
- PXE:
-
pseudoxanthoma elasticum
- SUR:
-
sulfonylurea-binding receptor
- TMD:
-
transmembrane domain
References
Armentano MF, Ostuni A, Infantino V, et al. (2008) Identification of a new splice variant of the human ABCC6 transporter. Biochem Res Int 2008:912478. doi:10.1155/2008/912478
Babenko AP, Bryan J (2002) SUR-dependent modulation of KATP channels by an N-terminal KIR6.2 peptide. defining intersubunit gating interactions. J Biol Chem 277:43997–44004. doi:10.1074/jbc.M208085200
Bakos E, Evers R, Calenda G, et al. (2000) Characterization of the amino-terminal regions in the human multidrug resistance protein (MRP1). J Cell Sci 113(Pt 24):4451–4461
Bakos E, Evers R, Szakács G, et al. (1998) Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J Biol Chem 273:32167–32175
Belinsky MG, Chen Z-S, Shchaveleva I, et al. (2002) Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6). Cancer Res 62:6172–6177
Biasini M, Bienert S, Waterhouse A, et al. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi:10.1093/nar/gku340
Birnbaumer L (2009) The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca(2+) concentrations. Annu Rev Pharmacol Toxicol 49:395–426. doi:10.1146/annurev.pharmtox.48.113006.094928
Boraldi F, Quaglino D, Croce MA, et al. (2003) Multidrug resistance protein-6 (MRP6) in human dermal fibroblasts. comparison between cells from normal subjects and from pseudoxanthoma elasticum patients. Matrix Biol 22:491–500
Chan KW, Zhang H, Logothetis DE (2003) N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J 22:3833–3843. doi:10.1093/emboj/cdg376
Chassaing N, Martin L, Calvas P, et al. (2005) Pseudoxanthoma elasticum: a clinical, pathophysiological and genetic update including 11 novel ABCC6 mutations. J Med Genet 42:881–892. doi:10.1136/jmg.2004.030171
Cuviello F, Tellgren-Roth Å, Lara P, et al. (2015) Membrane insertion and topology of the amino-terminal domain TMD0 of multidrug-resistance associated protein 6 (MRP6). FEBS Lett. doi:10.1016/j.febslet.2015.10.030
De Boussac H, Ratajewski M, Sachrajda I, et al. (2010) The ERK1/2-hepatocyte nuclear factor 4alpha axis regulates human ABCC6 gene expression in hepatocytes. J Biol Chem 285:22800–22808. doi:10.1074/jbc.M110.105593
Fang K, Csanády L, Chan KW (2006) The N-terminal transmembrane domain (TMD0) and a cytosolic linker (L0) of sulphonylurea receptor define the unique intrinsic gating of KATP channels. J Physiol 576:379–389. doi:10.1113/jphysiol.2006.112748
Finger RP, Charbel Issa P, Ladewig MS, et al. (2009) Pseudoxanthoma elasticum: genetics, clinical manifestations and therapeutic approaches. Surv Ophthalmol 54:272–285. doi:10.1016/j.survophthal.2008.12.006
Fiser A, Sali A (2003) Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol 374:461–491. doi:10.1016/S0076-6879(03)74020-8
Hegedus T, Orfi L, Seprodi A, et al. (2002) Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim Biophys Acta 1587:318–325
Higgins CF (1992) ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8:67–113. doi:10.1146/annurev.cb.08.110192.000435
Jansen RS, Küçükosmanoglu A, de Haas M, et al. (2013) ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci U S A 110:20206–20211. doi:10.1073/pnas.1319582110
Kamada K, Miyata M, Hirano T (2013) Molecular basis of SMC ATPase activation: role of internal structural changes of the regulatory subcomplex ScpAB. Structure 21:581–594. doi:10.1016/j.str.2013.02.016
Lee H, Lara P, Ostuni A, et al. (2014) Live-cell topology assessment of URG7, MRP6102 and SP-C using glycosylatable green fluorescent protein in mammalian cells. Biochem Biophys Res Commun 450:1587–1592. doi:10.1016/j.bbrc.2014.07.046
Mason DL, Michaelis S (2002) Requirement of the N-terminal extension for vacuolar trafficking and transport activity of yeast Ycf1p, an ATP-binding cassette transporter. Mol Biol Cell 13:4443–4455. doi:10.1091/mbc.E02-07-0405
Matsuzaki Y, Nakano A, Jiang Q-J, et al. (2005) Tissue-specific expression of the ABCC6 gene. J Invest Dermatol 125:900–905. doi:10.1111/j.0022-202X.2005.23897.x
Miglionico R, Armentano MF, Carmosino M, Salvia AM, Cuviello F, Bisaccia FOA (2014) Dysregulation of gene expression in ABCC6 knockdown HepG2 cells. Cell Mol Biol Lett 19:517–526
Mikhailov MV, Campbell JD, de Wet H, et al. (2005) 3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1. EMBO J 24:4166–4175. doi:10.1038/sj.emboj.7600877
Mikhailov MV, Mikhailova EA, Ashcroft SJ (2001) Molecular structure of the glibenclamide binding site of the beta-cell K(ATP) channel. FEBS Lett 499:154–160
Ostuni A, Lara P, Armentano MF, et al. (2013) The hepatitis B x antigen anti-apoptotic effector URG7 is localized to the endoplasmic reticulum membrane. FEBS Lett 587:3058–3062. doi:10.1016/j.febslet.2013.07.042
Ostuni A, Miglionico R, Monné M, et al. (2011) The nucleotide-binding domain 2 of the human transporter protein MRP6. J Bioenerg Biomembr 43:465–471
Paumi CM, Chuk M, Chevelev I, et al. (2008) Negative regulation of the yeast ABC transporter Ycf1p by phosphorylation within its N-terminal extension. J Biol Chem 283:27079–27088. doi:10.1074/jbc.M802569200
Sinkó E, Iliás A, Ujhelly O, et al. (2003) Subcellular localization and N-glycosylation of human ABCC6, expressed in MDCKII cells. Biochem Biophys Res Commun 308:263–269
Slot AJ, Molinski SV, Cole SPC (2011) Mammalian multidrug-resistance proteins (MRPs). Essays Biochem 50:179–207. doi:10.1042/bse0500179
Ter Beek J, Guskov A, Slotboom DJ (2014) Structural diversity of ABC transporters. J Gen Physiol 143:419–435. doi:10.1085/jgp.201411164
Uitto J, Pulkkinen L, Ringpfeil F (2001) Molecular genetics of pseudoxanthoma elasticum: a metabolic disorder at the environment-genome interface? Trends Mol Med 7:13–17
Volkov A, Mascarenhas J, Andrei-Selmer C, et al. (2003) A prokaryotic condensin/cohesin-like complex can actively compact chromosomes from a single position on the nucleoid and binds to DNA as a ring-like structure. Mol Cell Biol 23:5638–5650
Westlake CJ, Cole SPC, Deeley RG (2005) Role of the NH2-terminal membrane spanning domain of multidrug resistance protein 1/ABCC1 in protein processing and trafficking. Mol Biol Cell 16:2483–2492. doi:10.1091/mbc.E04-12-1113
Winkler M, Kühner P, Russ U, et al. (2012) Role of the amino-terminal transmembrane domain of sulfonylurea receptor SUR2B for coupling to K(IR)6.2, ligand binding, and oligomerization. Naunyn Schmiedeberg's Arch Pharmacol 385:287–298. doi:10.1007/s00210-011-0708-9
Xue P, Crum CM, Thibodeau PH (2014) Regulation of ABCC6 trafficking and stability by a conserved C-terminal PDZ-like sequence. PLoS one 9:e97360. doi:10.1371/journal.pone.0097360
Acknowledgments
We would like to thank the undergraduate student Michela Pupillo for her work on the homology model of L0 in ABCC6. This project was supported by the University of Basilicata.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
ESM 1
(PDF 2.55 mb)
Rights and permissions
About this article

Cite this article
Miglionico, R., Gerbino, A., Ostuni, A. et al. New insights into the roles of the N-terminal region of the ABCC6 transporter. J Bioenerg Biomembr 48, 259–267 (2016). https://doi.org/10.1007/s10863-016-9654-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10863-016-9654-z